Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
verify here. |
Cyberfriends: The help you're looking for is probably here.
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()

I [Allah] am nearer to you than the vein in your neck. --Koran
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- Blood Vessels
Lectures follow Textbook
QUIZBANK: Hemodynamic #'s 1-30, Vessels (all)
The term "arteriosclerosis" (literally, "hardening of the arteries") should be avoided by physicians. It includes (1) atherosclerosis; (2) Monckeberg's medial calcific sclerosis; and (3) arteriolar sclerosis (hyaline, hyperplastic, intimal fibrosis). Unqualified, the term usually means "atherosclerosis".
The vascular intima looks simple but isn't. Endothelial cells must maintain their no-stick inner surfaces, help constrict and dilate vessels, and heal damaged vessels. Myointimal cells and macrophages, located between the endothelium and the internal elastic membrane, are the principal actors in atherosclerosis.
Atherosclerosis is a stereotyped response of the inner surfaces of large arteries to a variety of insults. In this disease, the cells between the endothelium and the internal elastic membrane take up cholesterol-rich lipid, which then causes harm. Lesions progress from FATTY STREAKS to FIBROUS PLAQUES to COMPLICATED FIBROUS PLAQUES; they can also regress.
Atherosclerosis may calcify, but the problem in atherosclerosis is not the dystrophic calcification. Monckeberg's medial calcific sclerosis just means dystrophic calcification of the media of an artery, but it is almost never a real problem.
Atherosclerosis is an ancient disease, present in some Egyptian mummies (JAMA 302: 2091, 2009 -- the lesions are real but look quite mild by today's standards).
Atherosclerosis was THE great killer of 20th century North Americans. The epidemic peaked in 1968, and since then the decline has been spectacular, due more than anything else to healthier lifestyles (JAMA 277: 535, 1997; it's been steady since the mid-1980's: NEJM 339: 861, 1998). We are beginning to understand how the common risk factors relate to its pathogenesis. Americans are taking steps to protect themselves, and lifestyle changes can almost certainly reverse much of the damage in all but the most advanced lesions.
Hyaline arteriolar sclerosis results from damage to arterioles usually from increased pressure or increased blood glucose. Hyperplastic arteriolar sclerosis involves hyperplasia of the intimal cells; it results from processes that do severe, acute damage to the endothelium. Fibrosis of the intima results from high blood pressure or "just getting older."
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

William Harvey
OBJECTIVES: This is mastery material.
Recognize each of these anatomic lesions, grossly and/or microscopically as applicable:
Give a full account of the vascular changes contributing to, and resulting from, high blood pressure.
Give a full account of each of these clinical syndromes, with anatomic and clinical pathology as applicable:
Describe the range of infections that can affect the blood vessels.
Review the causes of deep vein thrombosis ("thrombophlebitis"), and describe its symptoms, signs, and principal complication.
Give accounts of each of the following, and be able to recognize them clinically:
Recognize each of these anatomic lesions, grossly and/or microscopically as applicable:
Describe the important tumors, hamartomas, and proliferations of blood and lymph vessels, how they behave clinically, and the pitfalls in making the diagnosis.
 NORMAL ANATOMY
NORMAL ANATOMY
ARTERIES carry blood from the heart.
ELASTIC ARTERIES ("large arteries") include the aorta and at least the beginnings of its largest branches. These arteries both propel and dampen the pulse wave. These are distinguished by a preponderance of elastic fibers in their media. The subendothelium of their intimal layers thickens over the course of life through the accumulation of collagen fibers and myointimal cells. The elastic tissue proliferates here. You already know that all elastic tissue slowly breaks down as we age. In older adults, the elastic is largely replaced by collagen. This also results in lengthening and thus tortuosity seen in older people. The adventitia and outer media are nourished by vasa vasora. All arteries depend on the blood within their lumens to nourish their intima and inner media.
MUSCULAR ARTERIES ("medium-sized arteries", "distributing arteries") exhibit smooth muscle in their walls, and may expand and contract to regulate the caliber of the lumen and thus the flow of blood. The intima is similar to that of the elastic arteries, and it thickens similarly. Smooth muscle may pass into the intima through fenestrae in the internal elastic membrane. These fenestrae may become wide in old age and be mistaken for damage from previous vasculitis. The media is bounded on either side by an inner and outer elastic membrane.
SMALL ARTERIES are the major site of autonomic regulation of blood flow, and take the worst beating in hypertension. A rule of thumb is that the wall and lumen should have the same thickness. Thickening of the intima occurs here as well. In sites of inflammation or tumor, it may be quite impressive (* "Friedlander's endarteritis obliterans", discovered by the real Dr. Friedlander, 19th century pathologist Carl, 1847-1887). Hyaline arteriolar sclerosis becomes a problem as we age, especially if we get diabetes or hypertension. There's no outer elastic membrane, and the layers become progressively less distinct as the arteries get smaller.
ARTERIOLES continue the anatomy of the small arteries. Two definitions that have been offered: (1) Arterioles have five or fewer layers of smooth muscle; (2) Arterioles have total diameter 100 microns or less.
VEINS and LYMPHATICS have histologic features that you know. In disease, veins do not usually show so much intimal proliferation and fibrosis as do arteries. The muscle in the wall of a vein is thinner, and in the larger veins tends to be less organized. Very large veins have some layers of elastic outside their muscular layer. Lymphatics run very close to arteries (even closer than the veins), and tend to be small and to have thinner walls than the vein that runs with that artery. It's not always possible to tell lymphatics from veins; if the vessel contains red cells, it's most likely a vein.
![]() Lymphatic vessels in edema
Lymphatic vessels in edema
They are not themselves plugged,
but are dilated from carring away fluid.
ENDOTHELIUM is special stuff. It is permeable to water and the small inorganic ions. It transports a little bit of blood protein by pinocytosis. Electron microscopists recognize it by the WEIBEL-PALADE BODIES (puh-LAH-dee, made of von Willebrand's factor). It can contract, to regulate capillary flow. It produces some of the subendothelial connective tissue. It also makes substances: (1) Prostacyclin (to keep its surface slippery); (2) Von Willebrand's factor; (3) Endothelin (a vasoconstrictor peptide); (4) Endothelial-derived relaxation factor (nitric oxide, EDRF; see Nature 368: 62, 1994).
VASCULAR SMOOTH MUSCLE is also special. It has LDL receptors. It can get into the intima through holes in the internal elastic membrane. Both facts will become important when we study atherogenesis.
 BIRTH DEFECTS INVOLVING VESSELS
BIRTH DEFECTS INVOLVING VESSELS
There are many variations on the normal anatomy of arteries and veins.
Malformations of the coronary arteries may first announce themselves by causing sudden death. One of the main coronary arteries may be absent or arise from the pulmonary artery. More about this soon.
The familiar red "birthmarks" are hemangiomas, and will be covered with "tumors".
The only other birth defects worth mentioning are BERRY ANEURYSMS and ARTERIOVENOUS MALFORMATIONS ("AV malformations", "AV fistulas", "AV aneurysms", etc.)
AV malformations involve a tangle of abnormal medium-sized vessels connecting a large artery and a large vein. The problem is shunting of the blood away from the territory that should be supplied by the artery. The vein will tend to expand ("aneurysm").
Sometimes the AV malformation is a mass of wormy vessels ("cirsoid aneurysm", "racemose aneurysm"; apparently endemic among Klingons). This is most common in the brain, where subarachnoid hemorrhage is the dread complication.
* Future pathologists: It's good to be able to distinguish a baby's AV malformation (which won't go away) from a baby's hemangioma (which probably will go away). An AV malformation does not stain for WT1; a hemangioma that will involute does stain for WT1 (Arch. Derm. 141: 1297, 2005).
* Genetic syndromes that affect vessels include Ehlers-Danlos type IV (the arterial walls become very weak and fragment), neurofibromatosis type I (lack of neurofibromin can cause aneurysms and malformations of the vessels especially in the head, or an artery may simply burst), and Fabry's (storage in the endothelium.)
Anomalies of the pulmonary veins (for example, some of them returning to the right atrium or superior vena cava) are part of the arcana of congenital heart disease -- subspecialist knowledge.
ATHEROSCLEROSIS (best recent update J. Am. Coll. Card. 46: 937, 2005; DeBakey
tries to sort out what puts you at risk for what Am. J. Card. 85: 1045, 2000)
![]()
{03476} coronary artery atherosclerosis
{06485} coronary artery atherosclerosis, mild
{06491} coronary artery atherosclerosis, severe
{06497} coronary artery atherosclerosis, total
{09446} êtat cribilé from multiple atheroemboli
|
|
Probably still the #1 killer of Americans.
Atherosclerosis causes harm by (1) narrowing / occluding arteries slowly over time (angina, sudden cardiac death without coronary occlusion, ischemic scarring of the myocardium, atherosclerotic dementia, leg claudication, intestinal angina, having one kidney waste away to a hypertension-producing nubbin); (2) occluding arteries suddenly by rupture of plaques (thrombosis, atheroembolization) or hemorrhages into plaques (myocardial infarct, atherosclerotic stroke, gangrene of the bowel); (3) weakening the walls of arteries (atherosclerotic aneurysms, penetrating atherosclerotic ulcers of the aorta).
* The radiologists are now able to see a fresh rupture of a fibrous cap, and correlating it with clinical findings: Circulation 105: 181, 2002; ultrasonography shows that, not surprisingly, the most common site is at the origin of the LAD (J. Am. Coll. Card. 46: 261, 2005).
* Your lecturer was once taken to task severely by opposing counsel's expert for describing "70% occlusion" of a coronary artery. Of course, it's really "70% narrowing", but as I pointed out, the New England Journal of Medicine has used the same language as your lecturer, and you'll see it often.
{06737} atherosclerosis of coronaries
{09443} atherosclerosis of middle cerebral artery
{10889} atheroembolus
You're already familiar with atheroembolization, which is a major cause both of stroke and of lower-extremity ischemia ("blue toes disease").
Atherosclerosis is a stereotyped response to injury featuring the accumulation of cholesterol-rich fat in the intima of the large and medium-sized arteries of the body. Typically these are phagocytes ("myointimal cells" -- smooth muscle cells that migrated from the media -- and macrophages derived ultimately from the blood though they proliferate in atherosclerotic lesions, even the early ones -- NEJM 369: 2352, 2013). These masses form PLAQUES, or ATHEROMAS.
The cholesterol-laden cells are of both macrophage (i.e., blood-monocyte) and medial-smooth-muscle origin. Today, the focus on "atherosclerosis as an inflammatory disease" has emphasized the role of macrophages, but both cell types are very much present.
For whatever reason, these cells have a great appetite for LDL cholesterol. When the LDL is abnormal (Ox-LDL, lipoprotein A; see below), or the LDL is processed down the "bad" (non-apoprotein B receptor-mediated) breakdown pathway, these cells accumulate lipid that damages them and their neighbors. The two pathways: Proc. Nat. Acad. Sci. 91: 4431, 1994.
A good working rule: The pulmonary arteries do not get serious atherosclerosis. Otherwise, if an artery has its own name, it can be significantly affected by atherosclerosis. The aortic surfaces of the aortic valve cusps are also prone to the disease at all stages (Heart 91: 576, 2005 documents what all pathologists already knew).
Atherosclerosis seems to be the stereotyped way in which most of the large named arteries respond to a variety of injuries. Don't expect a unified theory of atherogenesis anytime soon. However, the manageable risk factors are, in descending order (1-5 at least) of importance:
1. High levels of LDL cholesterol (LDL-C). This is the over-riding risk factor. Review Lancet 345: 362, 1995.
Your LDL level is a reflection of your heredity (no doubt), your diet (cholesterol-rich diets probably raise LDL some and the conventional wisdom is that saturated fatty acid-rich diets raise LDL -- more below), and your exercise habits (no doubt).
Our close non-human relatives and native hunter-gatherers run LDL-C levels at 50-70 mg/dL. There are contemporary hunter-gatherer peoples who do not get atherosclerosis, and if you can get a patient's LDL-C below 70, atherosclerosis predictably regresses (Med. Clin. N.A. 96: 13, 2012). By contrast, mummies from preindustrial civilizations (Egypt, Peru, southwest USA, and Aleuts) al have plenty of atherosclerosis (Lancet 381: 1211, 2013).
Also remember cholestatic jaundice, nephrotic syndrome, hypothyroidism (you need T3 to express LDL receptors on liver cells), amiodarone (* keeps T3 from letting you make LDL receptors) and (less striking) Cushingism.
As total cholesterol rises above 160 mg/dL, coronary risk rises roughly linearly.
* The Swedish simvastatin study, which laid to rest concerns about excess mortality from lowering LDL's: 345: 1274, 1995.
* By the way, don't try to sort out "saturated fat" as a risk factor for atherosclerosis. It's the received wisdom, but because "evidence from prospective studies has not supported a strong link between saturated fat intake and cardiovascular disease events", the nutrition folks at U. of Texas in Houston did a huge prospective study and found a weak risk from saturated fats in red meat and a weak protective effect from saturated fats in dairy (Am. J. Coll. Card. 96: 397, 2012).
2. Cigaret![]() smoking, "which damages the intima".
smoking, "which damages the intima".
* "Cigaret" is the preferred spelling. The suffix "-ette" makes the practice sound trivial and innocuous.
The old story about tobacco "damaging the intima" is giving way to the better-evidenced fact that tobacco smoke oxidizes LDL, making them into the poorly-digested form that accumulates in the intima.
Oxidized LDL is chemotactic for macrophages, makes them proliferate in the lesions, and is cytotoxic once they ingest it. It can make cells produce cytokines like α-TNF that may be fibrogenic, and there is even talk of antoantibodies.
Smoking oxidizes LDL, HDL protects it from oxidation, vitamin E prevents LDL oxidation (kind of, antioxidants seem to slow atherosclerosis JAMA 273: 1899, 1995), and so forth and so forth.
* Update on Ox-LDL in atherogenesis: J. Am. Coll. Card. 43: 1731, 2004. Superoxide plus NO. generates the noxious ONOO- peroxynitrite radical that oxidizes LDL (Proc. Nat. Acad. Sci. 91: 1044, 1994).
The claim that oxidized LDL is chemotactic for macrophgaes that become cholesterol-laden, and that it keeps them in the plaque, seems to be established (J. Clin. Inv. 119: 136, 2009; NEJM 360: 1144, 2009).
* You will probably run into the "pop" claim that "oxidized LDL is the cause of atherosclerosis and since there is very little oxygen in veins and in the pulmonary artery, this is why we do not see atherosclerosis here." If this were true, then (1) you'd see atherosclerosis in the smaller arteries, where oxygen tension is just as high; (2) in the pulmonary arteries of people with pulmonary hypertension, where the oxygen tension in the pulmonary artery is even less than in health though the pressure is higher, you'd see less rather than more atherosclerosis; (3) it's not true that there's very little oxygen in venous blood, especially if you're relaxing.
* A variety of other modifications of LDL also result in increased phagocytosis by macrophages, render it toxic to endothelium, smooth muscle, and macrophages, etc., etc. "Advanced lipoprotein testing" goes beyond LDL and HDL, to include non-HDL-cholesterol, apoB, LDL particle size, and other deep stuff. See Endo. Metab. Clin. N.A. 38: 1, 2009. One of many new risk factors is one's lipoprotein-associated phosopholipase A2, which correlates directly with risk: Lancet 375: 1498, 2010. It's not clear whether any of these will become "routine labs".
3. High blood pressure "which damages the intima". Review Lancet 345: 362, 1995.
4. Diabetes / longstanding hyperglycemia from any cause.
As a resident, I observed and was taught that when there is bad atherosclerosis in the smaller arteries (i.e., the ones without names in the anatomy books), there is usually diabetes. This is finally confirmed: Prog. Cardiovasc. Dis. 50: 112, 2007.
You'll read a tremendous amount about different effects of hyperglycemia, insulin, and so forth on lipoproteins, protein chemistry, and so forth. Some classic work (Science 258: 651, 1992) focuses on ADVANCED GLYCOSYLATION END-PRODUCTS ("glycation products"), proteins that have been non-enzymatically glycosylated (* Schiff-base --> Amadori product --> durable fluorescent product). Binding of these to endothelium makes it permeable, they inactivate nitric oxide, there are receptors for these substances which, when they bind, cause cells to produce fibrous tissue, glycosylated LDL probably can't be processed normally, etc., etc.
Once advanced glycation products have accumulated on board, runaway atherosclerosis affects even diabetics who have been restored to euglycemia.
* Another idea: High blood glucose levels cause overexpression of CD36, the LDL scavenger pathway receptor: Nat. Med. 7: 840, 2001.
| 5.
Lack of exercise (made "official" in 1992), independent of the above (NEJM
330: 1565, 1992).
The effects of physical exercise are numerous. Possibly "getting in shape" changes lipoprotein receptor counts, etc., etc. However, contrary to popular mythology, people who are very physically fit can and do still get serious atherosclerosis.
In the meantime, exercise capacity actually seems to be the best predictor of not dying of cardiovascular disease: NEJM 346: 793, 2002. And the famous step-two diet for improving LDL and HDL status actually fails in men and post-menopausal women if they refuse also to exercise: NEJM 339: 12, 1998. Despite some pop claims, the "big four" (cholesterol, smoking, hypertension, diabetes) still account for the vast majority of heart attacks: JAMA 290: 898, 2003. |
 |
6. A variety of biochemical lesions (hereditary, acquired) that promote thrombosis (NEJM 338: 79, 1998, lots more). Not yet "official", it is probably at least as important a risk factor as diabetes.
7. Other heredity: Important. Currently being sorted out as risk factors.
You are already familiar with the most common (80%) familial hypercholesterolemia, in which the LDL receptors (LDLR) are defective. Autosomal dominant; some people define "familial hypercholesterolemia" to be mutant LDLR. Over 1000 mutations are known.
You already are familiar with the second most common familial hypercholesterolemia, another autosomal dominant, in which there is a defective apoprotein B-100 receptor (this binds to the LDL receptor; again there are many different molecular variants). IDL's are not properly cleared by the liver and become excess LDL instead, and the "good" receptor-mediated pathway of LDL uptake by other cells is unavailable. This is called "familial defective apolipoprotein B100" if you want to get specific.
* Several other familial hypercholesterolemia syndromes are known. PCSK8 mutations cause LDL receptors to fall off the liver; LDLRAP1 (autosomal recessive) has something to do with the coated pits. Just watch these.
HDL is composed of A1 AND A2 APOLIPOPROTEINS.
It's been known for over a decade that transgenic mice with lots of A1 get much less atherosclerosis; those with lots of A2 paradoxically get more. Science 261: 469, 1993. A mutated apoA-1 that causes severe atherosclerosis in humans: J. Am. College Card. 44: 1429 2004.
* Insurers are now using various "complete" lipid profiles, including a directly-measured LDL plus HDL plus lipoprotein(a), remnant lipids (IDL and small VLDL), apo-B (a marker for the metabolic syndrome, suposedly), "small dense LDL" (a marker for the metabolic syndrome -- see Am. J. Clin. Path. 136: 20, 2011), "LDL pattern B". In 2008, the much-promoted screen was "VAP", a marketer's eponym.
FAMILIAL COMBINED HYPERLIPIDEMIA, i.e., high VLDL's, high LDL's, low HDL's ("my triglycerides and LDL both run high"), affects about 1% of folks and is a major coronary risk (3-10x increase over baseline). Patients are now considered a subgroup of the "syndrome X" family (more on this below) since most show central adiposity.
The illness is obviously polygenic. It is expressed best in adults. A mouse model has mutations at both apoprotein C-III and the LDL receptor (Science 275: 391, 1997). One human gene is apolipoprotein A2 (Nat. Gen. 18: 369, 1998.) Also watch cholesteryl ester transferring protein.
* LPR6 (LDL receptor protein 6) is definitely worth watching; severe mutants have elevated LDL and BP, diabetes, and coronary artery disease, plus accelerated osteoporosis (Science 315: 1278, 2007).
* "LIPOPROTEIN(A)" (Lp(a)) is apolipoprotein B100 bound to apolipoprotein(a). Levels seem to be mostly genetic, without dietary or environmental factors (J. Am. Coll. Card. 608: 716, 2012). These notes cited it a likely risk factor for atherosclerosis during the 1990's, and seems to have been forgotten for a while. By 2006, the recommendation was against routine measurement (JAMA 296: 1363, 2006); however, since it's now easy to measure, insurers are starting to screen ... and now it does seem to be a real risk factor (JAMA 301: 2331, 2009). Some folks treat an elevated level with niacin.
* Lipoprotein-associated phospholipase A2 (lp-pla2) is made by macrophages in plaques and is being used as a marker for atherosclerosis risk (JAMA 307: 2499, 2012).
* Common longevity mutation in cholesterol-ester transfer protein, which produces abnormally large LDL's and HDL's: JAMA 290: 2030, 2003; update JAMA 2008: 2777, 2008 (many alleles; correlation with vascular health is real but weak); NEJM 361: 2518, 2009 (two of the alleles are strong risks).
Over the past twenty years, there have been a host of reports of other putative genes placing one at risk for atherosclerosis. In 2007, a huge study (in Kansas City!) examined 86 putative genes at 70 loci and found only one (a fibrinogen variant) that was even nominally statistically significant (JAMA 297: 1511, 2007).
* American Heart Association on "genetics and genomics for prevention and treatment of cardiovascular disease": Circulation 115, 2878, 2007.
The GOOD news is that these genetic factors (at least whichever ones are real, as well as the havoc of hypertension) can be more or less neutralized by lowering LDL levels, by whatever means (Science 272: 629, 1996; Br. Med. J. 313: 1273, 1996).
8. LOW HDL AND ELEVATED FASTING TRIGLYCERIDES ("THE METABOLIC SYNDROME X").
Perhaps the HDL-C assay is most useful as a marker for the as-yet-poorly-understood "metabolic syndrome X" or "insulin resistance syndrome", with hypertension, insulin resistance, low HDL-C (less than 40 mg/dL for men, 50 mg/dL for women), high fasting triglycerides (150 mg/dL or more), fasting blood glucose over 100 mg/dL, some small, dense LDL particles that nobody understands, elevated ApoB that nobody understands, truncal obesity, low adiponectin (assays aren't any good yet), and a serious coronary risk even with normal LDL / total cholesterol.
This is a hot topic right now. Of course, the mainstay of therapy is diet and exercise (weight loss is key: Am. J. Card. 87: 827, 2001). Reviews Am. J. Epidem. 152: 897, 2000, Am. J. Card. 84(1A): 11J, 1999; Circulation 100: 123, 1999.
* Some fat in the liver, and elevated liver enzymes, is also typical: QJM 92: 73, 1999; today we call it "non-alcoholic fatty liver disease" (NAFLD). Many have the infamous "non-alcoholic steatohepatitis" (NASH) that we'll study later. NAFLD / NASH as independent risk factors for coronary artery atherosclerosis (how can you possibly control for hypertension / diabetes / inactivity): NEJM 363: 1341, 2010.
* No one understands why, but people with the metabolic syndrome also have more oxidized LDL: JAMA 299: 2287, 2008.
Watch for
* There is talk about nicotinic acid being superior to statins for the metabolic syndrome, but this does not seem to be affecting practice.
Remember that some of the antiretroviral therapies cause the metabolic syndrome (especialy, redistribution of fat); we may end up giving these people a growth-hormone releasing factor (NEJM 357: 2359, 2007).
* Dark chocolate as "an effective cardiovascular preventive strategy" in people with the metabolic syndrome: BMJ 344: e3657, 2012. Mmmm!!
* I predicted in the 1990's that obstructive sleep apnea would prove to be an independent risk factor and now there is support for this: Chest 140: 532, 2011.
* Rimonabant, the diet pill that blocks the cannabinoid type I receptor (the one that gives stoners the munchies) for the metabolic syndrome ("and it raises HDL-C and lowers glycosylated hemoglobin too!"): JAMA 299: 1547, 2008. It had to be withdrawn because people became depressed and suicidal.
9. Elevated levels of HOMOCYSTEINE.
Homocysteine can be elevated because of (1) an inborn error of metabolism, or (2) low intake of folic acid, or (3) low intake of vitamin B12. People with the bad inborn errors get horrendous atherosclerosis early in life.
Homocysteine, in turn...
There's now pretty good evidence that elevated blood homocysteine (as in folks who are deficient in folic acid, which is still probably true for many Americans) is a major independent risk factor for atherosclerosis (NEJM 332: 234, 1995; JAMA 281: 1817, 1999; Lancet 355: 523, 2000;) and a prospective series (JAMA 275: 1929, 1996). Review for everybody: Am. Fam. Phys. 56: 1607, 1997. Interest has waned over the past two decades.
Among the homocysteine-challenged.... Making early atherosclerosis shrivel with B6 and folic acid works: Lancet 355: 511, 2000. Supplementing all post-MI patients with vitamin B12 and folate reduces homocysteine and doesn't seem to hurt, but doesn't improve vascular outcomes (JAMA 303: 2486, 2010 -- darn).
11. Being on a protease inhibitor for HIV infection ("dyslipidemia": NEJM 356: 1723, 2007).
* 12. Having rheumatoid arthritis (damage to endothelium, "inflammation"; Arth. Rheum. 61: 1580, 2009).
13. Adult-onset growth-hormone deficiency. Now a robust finding. Still poorly-understood and hard-to-recognize, but supplementing seems to reverse the risk (J. Clin. Endo. Metab. 93: 3416, 2008).
NOTE: Despite all of the above, women are relatively protected from atherosclerosis until menopause. Afterwards, their risk increases rapidly to equal men's. Post-menopausal estrogen is protective (other bad press notwithstanding: Ann. Int. Med. 135: 939, 2001), and the addition of progesterone does not remove this protection: NEJM 335: 453, 1996.
NOTE: People of Japanese ancestry, living in Japan, have historically had relatively low risk for atherosclerosis. This has changed dramatically in Japan beginning in the early 1980's (Arch. Int. Med. 160: 2297, 2000). When they move to America and become Americanized, their risk approaches that of the rest of us.
NOTE: Finland and Scotland have slightly higher rates of atherosclerosis (reflected in rates of death from ischemic heart disease). The United States probably ranks next, followed by the rest of the "Western World", and far more than Japan or the poor nations. When a poor country develops rapidly, so does atherosclerosis (Atherosclerosis 153: 9, 2000).
NOTE: The French, with a very high-saturated-fat diet (that's part of why French cooking is so good), have a relatively low rate of atherosclerosis. This is probably due to their tremendous alcohol consumption; there was talk in the 1990's about tannins in red wine inhibiting oxidation of LDL's in the French. There could perhaps be something to this; watch resveratrol (an anti-oxidant from grapes) as an anti-atherosclerosis compound; the scandal over the best-known research group's papers broke in January 2012.
* NOTE: For the "chocolate is good for you" study (controlling for everything else, the more chocolate you eat, the less "cardiometabolic" disease) see BMJ 343: d4488, 2011.
* NOTE: Interest in carotenoids as atheroprotective has gotten little attention in the past decade despite some interesting animal work (Circulation 103: 2922, 2001.)
NOTE: The Inuit ("Eskimos"), who eat a lot of fat, are supposedly protected by omega-three fatty acids. These were promoted by pharmaceutical manufacturers as protective against atherosclerosis. The first refereed publication on the topic is an obscure French journal (Revue du Praticien 43: 164, 1993) which described the connection as a hypothesis and that "direct evidence through prospective preventive trials remains insufficient to establish that dietary supplements of marine oils prevent arterial diseases in humans efficiently." Omega-three fatty acids do have other effects, especially as anti-inflammatories, anti-platelet agents (in huge doses), and triglyceride-lowerers, but the mega-review in Mayo Clin. Proc. 75: 607, 2000 only concluded that they "may also be antiatherogenic." If you are confused by all the talk about recommending omega-3 polyunsaturated fatty acids after heart attack or for everyone, you will remain mystified after reading Lancet 376: 540, 2010. Less mystifying -- NEJM 367: 309, 2012, the "ORIGIN Trial" of grown-ups with glucose intolerance, a massive study in which omega-3's lowered triglycerides a tiny but statistically significant amount and did no good whatsoever as far as actually keeping anybody alive (this time, people with glucose tolerance problems and alive or healthy. "Treat the person, not the lab value" -- ERF Omega-3 fatty acid supplementation was a total failure in reducing the risk of every major cardiovascular disease event: JAMA 308: 1024, 2012. A total failure in folks with multiple risk factors: NEJM 368: 1800, 2013. So far as "the Eskimo paradox" goes, there has been essentially nothing written since the early 1990's -- the two recent population studies found no correlation between omega-3 fatty acid levels and atherosclerosis (which in fact now is worse among the Eskimos than other folks -- Stroke 39: 3079, 2008), and that body levels of omega-3 fatty acids has no effect on the extent of atherosclerosis, and the relationship between high intake and less intimal-medial thickness is weak (Atherosclerosis 199: 346, 2008). The Inuit / Eskimos are the shortest-lived, sickliest people in the Western Hemisphere. You can read about their terrible health problems (notably alcoholism and malnutrition) on your own; the government of Canada is finally on the right track by distributing free fruits and vegetables to these unfortunates.
NOTE: "No-cholesterol" dietary items are still maybe LDL-raisers and supposedly atherogenic if rich in saturated fat.
NOTE: After 1991, the coronary artery mortality in Poland plummeted. Probably it's from a better diet, with more fresh fruits and vegetables and vegetable fats and less lard (BMJ 316: 1047, 1998).
NOTE: You can go nuts trying to keep track of which factors and their remediation affect, correlate with, and "are associated with" which other factors, listed or rumored to be important in atherogenesis. I am unpersuaded that "stress", independent of these other variables, is a significant risk factor for atherosclerosis. (For a review of the claim that the hormones produced by psychological stress slowly promote atherosclerosis, see Lancet 370: 1089, 2007; for the London study on job stress which concluded the relationship was real but weak, see Lancet 380: 1491, 2012). However, higher serum epinephrine levels probably would increase a person's risk of sudden cardiac death in a potentially rhythm-disturbing situation, all other things being equal.
* NOTE: You may still read about the "type A behavior pattern" (variably described as high-achieving, ambitious, competitive, work-driven, time-urgent, hostile, or whatever) causing atherosclerosis. From tobacco company documents, we now know this claim was nothing more than a cynical Philip Morris company attempt to divert attention from the dangers of tobacco. See Am. J. Pub. Health 102: 2018, 2012.
NOTE: I am still not aware of any reason to believe that obesity is a major risk factor for atherosclerosis except insofar as it is related to the above (i.e., high cholesterol from bad diet, smoking, hypertension, diabetes, lack of exercise, metabolic syndrome X). Obesity (especially "central obesity" / "syndrome X type", i.e., the dude's butt-crack shows over his beltline when he squats) produces the hormone resistin, which renders muscle and liver resistant to the effects of insulin and maybe does other things. Watch this one.
* NOTE: Don't ask me to sort out the "trans-fatty acids are evil" prevailing wisdom. The original synthetic trans-fatty acid was Crisco; however, there are a few natural trans-unsaturated fatty acids as well. The huge review in NEJM 354: 1601, 2006 is filled with multivariate stuff on people and effects on molecules, but strangely there is only a single animal experiment, only marginally relevant, cited. Animal models remain conspicuously few. In the only one that involves atherosclerosis, the atherosclerotic lesions were "variable" (Br. J. Nutr. 102: 1811, 2009). A group in India reported increased muscle insulin resistance in rats but failed to find the hepatic steatosis or vascular lesions that the trans-fats are supposed to produce (Ann. Nutr. Metab. 58: 272, 2011). A ten-rat study from an obscure Japanese medical school in an obscure journal found nothing striking: J. Oleo Science 62: 57, 2013 -- some liver changes was all. The business is emotional and obviously politicized. New York City banned trans-fats from restaurant food (Ann. Int. Med. 151: 129, 2009 -- a diner wouldn't be able to tell any difference.) The authors' claim that trans-fatty acids cause 30,000 premature deaths in the USA each year seems to come from thin air, as it does NOT appear in the NEJM article to which it is referenced but is extrapolated from the assumption that because somebody's group of Crisco-eaters had 30% higher C-reactive protein.... well, you can understand how difficult it is to sort everything out. The much-discussed ongoing Nurses' Health Study could find an impressive correlation between red-cell trans-fatty acid concentration and atherosclerosis only in the upper quartile and didn't control for such obvious things as obesity and lack of exercise; to be fair, there was a positive correlation between total LDL cholesterol and trans-fatty acid levels (Circulation 115: 1858, 2007). The one real prospective study shows no impact on sudden cardiac death in nurses (Am. Heart J. 158: 761, 2009). The recent big French study couldn't find any clear effect of either synthetic or natural trans-fats on lipid profiles or cardiovascular mortality in ordinary, non-hyperlipidemic folks (Am. J. Clin. Nutr. 87: 558, 2008.) Am. J. Clin. Nutr. 90: 88, 2009 refuted the claim that trans-fats impair insulin sensitivity in overweight ladies. Underclass kids have "reduced dietary quality" including eating a lot of trans-unsaturated fats (surprise! J. Am. Diet. Assoc. 109: 1612, 2009) and overall fats as well. Go figure. Given that gorging on Crisco-cooked french-fries may run with other unhealthy habits, sorting everything out is impossibile -- thankfully, nobody will miss trans fats except Crisco stockholders. The latest review acknowledges that the case against trans-fats is based on epidemiology without any known mechanism: Mol. Nutr. Food Res. 56: 1090, 2012. For "trans-palmitoleic acid", which occurs in dairy and is now reported as being good for people's vascular health, see Am. J. Clin. Nutr. 97: 854, 2013.
NOTE: I have never been impressed with isolated fasting triglyceride levels as an independent risk factor (neither was the NIH consensus panel JAMA 269: 505, 1993; and twenty years later the subject is still confusing, with no clear role for this easy-to-measure lab value Med. Clin. N.A. 96: 39, 2012). Iron overload may also be a risk factor maybe by oxidizing LDL (yes! Circulation 96: 3300, 1997; no! NEJM 330: 1119, 1994), the no's seem to have won. Claims about coffee has repeatedly flopped; in the most recent study, drinking either the real stuff or decaf actually seems to protect (Am. J. Med. 148: 904, 2008). The "baldness is an independent risk factor" article (JAMA 269: 998, 1993; even a bright kid could point out methodologic howlers) was sponsored by the hair-restorer folks anticipating some lawyer finding "a higher rate of MI's in people using hair-restorer".
NOTE: Consensus panel on lipid profiling (JAMA 269: 505, 1993) recommends screening HDL-C along with routine cholesterol screens, but can't find much reason to check triglycerides or consider them an independent risk factor.
* NOTE: On November 30, 2010, Uncle Sam declared that "VA presumes Veterans' ischemic heart disease as associated with [sic.] exposure to Agent Orange or other herbicides during military service. Vietnam-era Veterans exposed to herbicides do not have to prove a connection between their ischemic heart disease and military service to be eligible to receive VA benefits." The evidence in peer-reviewed publications? A study of carotid artery thickness in a whopping eleven Agent Orange factory workers from the '60's (Neuroendo. Lett. 30 S1: 219, 2009). Two mortality studies among factory workers showing no effect (J. Occup. Env. Med. 51: 1049 and 1212, 2009). An NIH-supported review of all the published work (mostly on factory workers rather than soldiers) that ended up concluding that there seemed to be a link but there were way too many confounding variables (Env. Health Perspect 116: 1443, 2008). A 10% increase in coronary heart disease in a restrospective study of dioxin factory workers (Am. J. Epidem. 170: 501, 2009). A mouse study on lipoproteins (no heart attacks in the mice, but dioxin was toxic to cells in tissue culture -- no surprise) -- Card. Tox. 1: 285: 2001. A study with no attempt to rule out recall bias: Env. Health Perspect 106(S2): 645, 1998 -- supplements like this tend not to be peer-reviewed. Everything else predates 1996. What is obviously happening is that science is taking a back seat to politics, and that as Vietnam veterans reach retirement age and start suffering in large numbers from angina and heart attacks, they can be compensated and cared for like other older people. Further, having the VA take over the care for this usually-very-manageable condition will probably make compliance easier, and ultimately save a lot of money and keep these people healthier. Your lecturer certainly does not have a problem with doing something extra to help our Vietnam veterans, good science or no.
* CONTROVERSY: "Atherosclerosis as an infectious / inflammatory disease."
From time to time, people find what seem to be bits of microbes in atherosclerotic plaques and suggest the bugs are etiologic. DNA sequences from various bugs get reported fairly often in plaque debris. Immunofluorescence identifies antigens from these bugs in the debris as well. Since many different "microbes" have been identified in this way (including the very unlikely Helicobacter pylori), and since the bacteria themselves are never convincingly visualized, the obvious explanation is that the plaque grunge itself soaks up whatever microbial debris may be in the bloodstream.
The CMV![]() -causes-atherosclerosis flap of the 1980's came to nothing.
In the 1990's it was
chlamydia-in-your-plaques (J. Inf. Dis. 175: 883, 1997;
negative study Br. Med. J. 318: 1035, 1999, J. Am. Coll. Card. 41: 1482, 2003;
two more Br. Med. J. 321: 204 & 208, 2000).
Even the proponents admit the case is hardly made: JAMA 288: 2724, 2002. The inflammation
in plaques hardly resembles what you see in
trachoma
-causes-atherosclerosis flap of the 1980's came to nothing.
In the 1990's it was
chlamydia-in-your-plaques (J. Inf. Dis. 175: 883, 1997;
negative study Br. Med. J. 318: 1035, 1999, J. Am. Coll. Card. 41: 1482, 2003;
two more Br. Med. J. 321: 204 & 208, 2000).
Even the proponents admit the case is hardly made: JAMA 288: 2724, 2002. The inflammation
in plaques hardly resembles what you see in
trachoma![]() ,
psittacosis
,
psittacosis![]() ,
lymphogranuloma venereum
,
lymphogranuloma venereum![]() or other known chlamydial illnesses -- these display spectacular proliferation
of lymphoid germinal centers and abundant plasma cells, and/or suppurating
granulomas.
Chlamydia are easy to see in the illnesses they obviously cause,
but in atherosclerosis the actual organisms (rather than just their DNA
or antigens) remain incredibly elusive.
The most recent sighting is Acta. Biol. Hung. 86:
233, 2005 and the "chlamydia" were calcified and of varying sizes ... just as
in classic dystrophic calcification without any organisms.
Draw your own conclusions.
or other known chlamydial illnesses -- these display spectacular proliferation
of lymphoid germinal centers and abundant plasma cells, and/or suppurating
granulomas.
Chlamydia are easy to see in the illnesses they obviously cause,
but in atherosclerosis the actual organisms (rather than just their DNA
or antigens) remain incredibly elusive.
The most recent sighting is Acta. Biol. Hung. 86:
233, 2005 and the "chlamydia" were calcified and of varying sizes ... just as
in classic dystrophic calcification without any organisms.
Draw your own conclusions.
Correlating the presence of a microbe and severity of atherosclerosis makes for an extremely easy "scientific study." And these studies are driven by the hope that atherosclerosis might respond to an antibiotic-based regimen as has worked so well for stomach ulcers. A key difference is that everybody can see the bacteria in stomach ulcers, and nobody can actually see them in atherosclerosis. And a conspicuous lack of controls distinguishes most of the "positive" studies I've seen. For example, a handful of studies finding that people with helicobacter in their stomachs get worse coronary atherosclerosis fail to control for tobacco smoking and dietary habits. (We'll let you find this stuff on your own.)
Most recently the talk is about "infectious burden", i.e., the more different usual suspects that you're
making antibodies against, the greater your risk of dying of a heart attack
(Circulation 105: 15, 2002). The fact that the "usual suspects" now
include genital herpes![]() and stomach helicobacter invites the idea that
old folks who are sicker are more likely to have polyclonal B-cell activation, which is simply
common sense.
and stomach helicobacter invites the idea that
old folks who are sicker are more likely to have polyclonal B-cell activation, which is simply
common sense.
Update on "pathogens and atherosclerosis": Thrombosis and Hemostasis 106: 858, 2011. The authors share my skepticism and note that antimicrobials for atherosclerosis have been a total failure. The most recent article on infection as the cause of atherosclerosis has an elaborate hypothesis about immune complexes and such but no pathology to support it (Am. J. Med. Sci. 344: 391, 2012).
C-REACTIVE PROTEIN is still the subject of much excitement, since serum levels in the absence of the acute phase reaction seem to correlate with the extent of atherosclerosis in both humans and animal models of atherosclerosis.
There's much talk about the "proinflammatory phenotype" (J. Clin. Endo. Metab. 90: 4549, 2005), i.e., the person who's likely to have high C-reactive protein levels in the absence of obvious inflammation. Surprise! These people are older and fatter, have higher triglyceride levels, higher insulin levels (i.e., insulin resistance), and higher leptin levels -- all obviously involved with atherosclerosis. For some reason, other acute-phase-reaction markers (fibrinogen, stimulated interleukin beta, some more obscure proteins) also average higher. People with a mutation that elevates C-reactive protein are not at extra risk for atherosclerosis (told you so.... NEJM 359: 1897, 2008).
All this says to me is that there is some link between the C-reactive protein system and lipid metabolism. The conclusion that people with atherosclerosis harbor more smoldering infections hardly seems warranted.
Update on the molecules that are supposedly involved in "inflammation as a cause of atherosclerosis": Circulation 109(S2):II-2, 2004. I'm anything but impressed by the "lymphocytic infiltrates" that supposedly abound in plaques, especially as they get worse. Somebody else will tell you that "inflammation is usually what causes the plaques to rupture", which is hard to believe given their strong propensity to rupture at times of hard physical or emotional exertion -- a mechanical explanation would seem easier to believe.
An enormous study of the impact of adding C-reactive protein and fibringen to the prognostic model that already includes age, sex, smoking status, blood pressure, history of diabetes and total cholesterol suggests that as independent risk factors, their value is probably real but very small (NEJM 367: 1310, 2012).
Supporting your lecturer's skepticism is a single study that addressed the obvious question. Mice with a genetic predisposition to atherosclerosis get exactly the same lesions whether they are germ-and-virus free or have the usual microflora (J. Exp. Med. 191: 1437, 2000.) No one has tried to challenge this work, and this fact alone tells me something about the whole field. There's been little talk about this stuff in the past decade.
NOTE: There are several situations in which development of atherosclerosis is ACCELERATED. These include
In these situations, "endothelial damage" and "thrombosis" appear to be critical. Still helpful: Mayo Clin. Proc. 66: 818, 1991. Okay, atherosclerosis is actually a pattern of injury.
{03473} atherosclerosis, in a vein graft
{06581} atherosclerosis after radiation
The innocent precursors: (anatomy: Am. J. Path. 143: 1444, 1993)
{11057} fatty streak
{25024} fatty streak
{41533} foam cells
Don't worry about FATTY DOTS, tiny groups of macrophages with cholesterol. Every toddler in the world probably has FATTY STREAKS, masses of lipid-rich foam cells (mostly macrophages as this stage) in the intima. These impart a pretty two-tone appearance to the intima of most everyone's aorta, without causing other problems. Most researchers now consider these precursor lesions to common-type serious atherosclerosis.
Not yet explained: (1) Their distribution differs from serious plaques; (2) Their presence seems unrelated to diet, etc., (3) They seem to regress later in life as fibrous plaques grow more severe.
We've got plenty still to learn about the pathogenesis of the fatty streak, but parts of the picture have recently become clear.
Fatty streaks result from macrophages taking up LDL cholesterol, but the LDL (we think) must first be altered to OXIDIZED LDL ("Ox-LDL", i.e., with its cholesterol and/or unsaturated fat peroxidated) taken up by an exotic "Ox-LDL receptor" (cloned Nature 385: 73, 1997) on the macrophage surface, and processed via the "scavenger pathway". The alteration can be brought about by endothelium or smooth muscle.
The risk factors have little to do with fatty streaking, which is universal. Boys have more than girls, blacks more than whites, it takes over maybe 30% of the surface by the teens.
The time bombs: Fibrous plaques
{11054} fibrous plaques
Later in life (and it all depends on risk factors), a person develops FIBROUS PLAQUES. These are masses of cholesterol-rich cells with an overlying FIBROUS CAP, a mass of dense collagen with some elastin as well. There may be dystrophic CALCIFICATIONS, extensive fibrosis and/or smooth muscle hyperplasia in the plaque, and some calcification. Cholesterol-laden cells appear as FOAM CELLS; these are usually both of smooth muscle and macrophage origin. When they die, there will be CHOLESTEROL NEEDLES (remember these?) and GRUMOUS DEBRIS. ("Grumous" is a charming near-synonym for "grunge", i.e., granular, semi-solid material.)
* More marvels: A MRI contrast medium that lights up only elastin, showing plaques (Nat. Med. 17: 383, 2011).
In acute coronary occlusion, angioplasty fails to establish re-flow in up to 30% of cases. We now know that many of these people actually have the grunge ("plaque gruel") released from the plaques by the procedure, and this itself plugs the artery (Circulation 106: 1672, 2002).
* While we are talking about calcium... "Chelation therapy" is an old "alternative treatment" for atherosclerosis. The patient is given a calcium chelator, usually EDTA, by vein, and told that this removes the plaques, which are supposedly made primarily from calcium. This causes tingling of the fingers (transient hypocalcemia) which the practitioner tell the patient is due to improved circulation to the fingers. No need to belabor why this is nonsense, and of course the empirical work has shown no effect on plaques. "Chelation therapists" promote healthy lifestyles and perhaps they serve a useful function for this reason. Anyone practicing "chelation therapy" nowadays is knowingly deceiving the public.
In at least many cases, atherosclerotic lesions are monoclonal, suggesting that Nowell's law / natural selection operates in their pathogenesis. This isn't surprising, especially since (1) we now know that oxidized-LDL is mitogenic for macrophages, and (2) the smooth muscle of the aorta itself is made of large groups of clonal cells (Am. J. Path. 152: 913, 1998). However, the old claim that each plaque arises from a single cell has now been amply refuted.
Much work now indicates that progression from the fatty streak to the fibrous plaque is at least in part the result of incorporation and organization of thrombi (* "the Rokitansky theory", makes sense). Most fibrous plaques are rich in material that immunostains as fibrin (though this is absent from normal artery or fatty streaks).
The distribution of the fibrous plaques is fairly predictable. Probably because of increasing turbulence (farther downstream, and running up against the iliac bifurcation), they increase centrifugally in the aorta. They are especially common at bifurcations "where turbulence damages the endothelium". Unfortunately, the carotid bifurcation and origin of the left anterior descending coronary artery are favorite sites. Renal artery ostia are vulnerable, though the rest of the renal artery is spared. Iliac arteries get it much worse than brachial arteries. The low-pressure pulmonary arteries are almost never seriously affected; finding any plaques at all strongly suggests pulmonary hypertension.
The killers: Complicated plaques
{11060} the nasty stuff
{14216} the nasty stuff
{25461} the nasty stuff
{53265} the nasty stuff
![]() Atherosclerosis
Atherosclerosis
Prosthesis in place
Urbana Atlas of Pathology
As a person continues to be at risk for atherosclerosis, the plaques become COMPLICATED. Any of the following unpleasant things can happen:
Platelets, fibrin, or both accumulate wherever the endothelial cells are damaged or lost. Platelets produce such factors as platelet-derived growth factor, transforming growth-factor beta, and others. Fibrin makes endothelial cells migrate (rendering the surface permeable), causes vascular smooth muscle to proliferate, presents a surface on which LDL may accumulate, binds the LDL variant Lp(a) (i.e., LDL with a bound apoprotein A molecule) that accumulates in the plaque with the fibrin, etc., etc. Fibrin degradation products also enhance vascular permeability.
There are three different lesions that can be seen beneath a coronary artery thrombus (J. Am. Coll. Card. 47: C13, 2006).
The thrombus can embolize if the artery is large, or plug the lumen if the artery is small (i.e., a coronary or cerebral artery). This is probably the cause of a majority of myocardial infarcts and cases of unstable angina pectoris.
If the "Rokitansky theory" is correct, the thrombus is even more likely to become incorporated into the plaque, making it grow. For an update on thrombus formation on top of plaques, see Ann. Int. Med. 134: 224, 2001.
ENDOTHELIAL DAMAGE ALONE might result from "high LDL" (see above), a virus (seems doubtful), or shear forces of flowing blood (plausible, since atherosclerosis is worse at bifurcations). As noted, the body responds by accumulating lipids and monocytes with cell-derived growth factors causing smooth muscle cells to proliferate. This probably begins the spontaneous atherosclerosis process.
CRACKING-FISSURING OF THE FIBROUS CAP (old review: Circulation 82(S II): II-47, 1990) leads to deposition of at least a platelet layer, and often a real thrombus. There are often cracks in coronary arteries and little thrombi even in patients dying of something other than MI.
If the injury reaches the MEDIA, the clotting cascade will surely be activated, smooth muscle cells will migrate in, and fibrosis will occur. And thrombin appears to be mitogenic for smooth muscles cells (either directly or indirectly).
Since atherosclerosis is a patchy process, it would seem reasonable to think that plaques exacerbate local conditions that contribute to their own growth. Two such mechanisms could be thrombosis and turbulent blood flow.
This is probably the second most common mechanism by which a coronary artery becomes occluded.
{06524} lethal hemorrhage into a plaque in a coronary artery
![]() Hemorrhage into a plaque
Hemorrhage into a plaque
Caused death
ERF/KCUMB
The good news: It's reversible
In the regressed lesion, the intracellular lipid vanishes, the extracellular lipid where the cells have died diminishes, the fibrous cap remodels and flattens, and the endothelial damage overlying the fibrous cap heals. See Am. J. Card. 65: 33F, March. 20, 1990. Not surprisingly, the reallly hard, calcified lesions are least likely to regress (J. Am. Coll. Card. 49: 263, 2007).
For an update on plaque regression using statins, see J. Am. Coll. Card. 46: 106, 2005.
A similar, huge study: Am. J. Med. 118(S-12A): 22, 2005.
If you believe the "inflammation" model of atherosclerosis,
they you'll accept that "statins are the most effective
agents available today for the reduction of vascular inflammation (Circulation 109(S2):II-2, 2004 --
they do rapidly lower C-reactive protein levels independent of LDL levels, but
of course, they're not front-line for other inflammatory diseases);
whatever you decide, the statins do stop macrophages from making their own cholesterol.
This is now mainstream;
I predicted this in 1994.
* The "Pathobiological Determinants of Atherosclerosis in Youth Study"
(JAMA 281: 727, 1999) looked at people under age 35
who died of all causes
and found fatty streaks to be ubiquitous and
little fibrous plaques fairly common. Any pathologist could have showed you without
your having to spend any money, Uncle Sam. Accepting the study's conclusion
that "primary prevention of atherosclerosis must begin in childhood
or adolescence" requires us to make the dubious assumption that
today's harmless streak or bump
must be tomorrow's killer plaque, and that today's normal-histology-book
coronary is far more likely to be clean in 30 years. Given the
suddenness with which severe atherosclerotic lesions appear (consider
the effect of female menopause) and regress (see below), that's
asking us to assume a lot.
THINK! In the space below, review how each risk factor or intervention might work at the molecular and chemical levels:
High LDL is bad
Cigaret smoking is bad
High blood pressure is bad
Diabetes is bad
An aspirin a day is good (HINT: Thrombi and platelets, maybe "reduced inflammation")
NOTE: Don't confuse atherosclerosis with the notable non-disease, MONCKEBERG'S MEDIAL CALCIFIC SCLEROSIS (see below).
NOTE: Together, atherosclerosis, Monckeberg's, and the two kinds of arteriolar sclerosis listed below are ARTERIOSCLEROSIS. In other words, avoid using that word altogether.
* NOTE: When diet and exercise fail....
Cholestyramine / colestipol: Binds cholesterol in the gut
Niacin: Decreased LDL and VLDL synthesis (always popular; no clinical benefit when given with statins JAMA NEJM 365: 2255, 2011).
Gemfibrozil: Enhances VLDL clearance
Statin family: Blocks cholesterol synthesis
* Torcetrapib, the failed wonder drug that inhibited cholesterol ester transfer protein, decreased LDL and increased HDL marvellously, but had no effect whatsoever on atherosclerosis, actually increasing "cardiovascular events". Puzzle that out. See NEJM 356: 1302, 2007. Probably this was because the HDL's weren't accept cholesterol from atheromas ("cholesterol efflux capacity" -- explained NEJM 364: 127, 2011). Perhaps anacetrapib will work better: NEJM 363: 2406, 2010.
MONCKEBERG'S MEDIAL CALCIFIC SCLEROSIS
|
|
Dystrophic calcification (sometimes even ossification) of the media of arteries, typically in older adults.
This is a common, banal, pretty much harmless process. At worst, it can widen one's pulse pressure due to decreased aortic compliance, or make "radial artery blood-gas sticks" difficult and hazardous. It may be visible on x-ray.
Despite the term "medial", the internal elastic membrane is always calcified (settled Arch. Path. Lab. Med. 132: 47, 2008).
* Evidently the smooth muscle cells produce at least four proteins that indicate they want to make bone. You can read about it in Circ. 100: 2168, 1999.
ARTERIOLAR SCLEROSIS ("arteriolosclerosis")
|
|
|
Three processes that narrow the lumens of the small arteries and arterioles in some or all of the body
INTIMAL FIBROSIS or (better) "fibroelastic hyperplasia" (and some call it "intimal sclerosis"), is the slow buildup of fibrous tissue (usually with some layers of elastic) in the intima of a small artery. It's a part of aging, and is exacerbated by high blood pressure. It involves smaller arteries (rather than the larger ones, as in atherosclerosis) and doesn't feature the lipid buildup. It also spares the arterioles. The most familiar form is the damage to the small arteries of the kidney seen in longstanding high blood pressure.
HYALINE ARTERIOLAR SCLEROSIS: slow buildup of basement-membrane type material, eventually obliterating the cellular structure of the wall and narrowing the lumen. Mostly the arterioles are involved.
The most common place where we see this is in the arterioles of the kidney in high blood pressure and diabetes.
However, it turns up often, and can appear anywhere. The most familiar causes are
No one really knows why any of these causes "hyaline arteriolosclerosis", but the anatomic pathology is impressive. Hypertension is the least potent. The effects on tissue perfusion, and probably on the sympathetic regulation of blood pressure, are not salutary.
Future pathologists: Look in the fat just outside the adrenal capsules to get a good idea about the extent of systemic hyaline arteriolosclerosis. Lots of hyaline change of the small arteries here is a great marker for longstanding hypertension. "Binswanger's encephalopathy" is a dread, Alzheimer-like dementia caused by hyaline arteriolosclerosis of the brain.
* If you like, you can consider amyloidosis of small arteries, which hyalinizes but is less likely to cause much stenosis, to be another cause of hyaline arteriolar sclerosis. No one will argue or care.
{11777} hyaline arteriolar sclerosis
{40267} hyaline arteriolar sclerosis
{40347} hyaline arteriolar sclerosis

HYPERPLASTIC ARTERIOLAR SCLEROSIS: Concentric, often rapid proliferation of the intimal (or sometimes smooth muscle) cells of an arteriole. "Onion-skin arteriole".
Fortunately uncommon. The causes are
{39559} hyperplastic arteriolar sclerosis in pulmonary hypertension
{24854} hyperplastic arteriolar sclerosis, scleroderma kidney
|
|
Note that all these are processes that specifically damage the intima of vessels, which presumably undergo hyperplasia in response. This is a much more aggressive process. (You could think of it as a callus of the endothelium.)
Note that any of these processes can be severe enough to cause some necrosis of the vessel. Malignant hypertension is usually this severe.
Don't confuse either of these processes with the inexorable FIBROUS THICKENING OF THE INTIMA OF SMALL ARTERIES in mild hypertension and normal aging. This is so common and so important that IT DOESN'T HAVE A NAME BEYOND "INTIMAL FIBROSIS".
TRANSPLANT VASCULOPATHY is a concentric fibrous thickening, mostly confined to the intima, in allografts (heart, kidney, liver, others) that have survived for a long time; the process develops rather abruptly, and while immunity must be a factor, it cannot be the only explanation. See Am. Heart. J. 129: 791, 1995.
THE VASCULITIS FAMILY (update Am. J. Clin. Path. 124(S): S-84, 2005)
|
|
|
Scarring can narrow a vessel, or cause an artery to balloon (aneurysm).
An inflamed vessel may rupture (petechiae, purpura).
POLYARTERITIS NODOSA
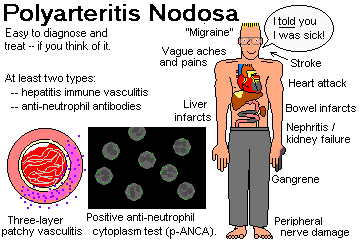
Due to hepatitis B infection (antigen-antibody complexes), anti-myeloperoxidase disease ("anti-MPO", "anti-neutrophil cytoplasmic antibody disease", now distinguished from "true polyarteritis nodosa"), or "idiopathic".
The principal discussion of polyarteritis nodosa, Wegener's, and their family comes under immunopathology. Remember classic polyarteritis nodosa as an important cause of infarcts anywhere in the body except lung.
LEUKOCYTOCLASTIC VASCULITIS
Generally type III immune injury of the venules, often diagnosed on skin biopsy (the patient has "palpable purpura"). Common, but infarcts and serious damage are fortunately rare.
"Leukocytoclastic" refers to the dead neutrophils lying about, visible as nuclear dust.
{14284} leukocytoclastic vasculitis
{14286} leukocytoclastic vasculitis
{14287} leukocytoclastic vasculitis
{14289} leukocytoclastic vasculitis
{14290} leukocytoclastic vasculitis
{14292} leukocytoclastic vasculitis
{14293} leukocytoclastic vasculitis
{14294} leukocytoclastic vasculitis
{14295} leukocytoclastic vasculitis
{14296} leukocytoclastic vasculitis
{14298} leukocytoclastic vasculitis
Mostly this results from taking medicines. Less common causes are cryoglobulinemia (how?), lupus and its kindred, and the antigenemia of HBV and malignancy.
WEGENER'S GRANULOMATOSIS
A vasculitis, usually with granulomas, caused by an anti-proteinase 3 autoantibody (anti-PR3). Lung cavities, segmental necrotizing glomerulonephritis with crescents, and/or vanishing nose, all most likely with granulomas. We'll talk more about this in "immuno".
CHURG-STRAUSS DISEASE
{38497} old burned-out Wegener's, without good granulomas
TEMPORAL ARTERITIS ("cranial giant cell arteritis", when elsewhere "giant cell arteritis")
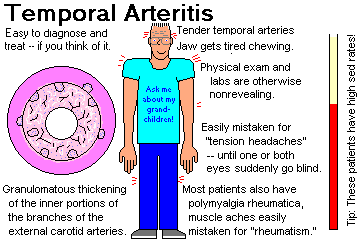
A disease of older folks, mostly over 60 (nobody's immune), still of unknown etiology, in which the macrophages seem to become angry with the internal elastic membrane of the arteries of the external carotid system.
"Jaw claudication" (tired jaw on chewing) is a picturesque syndrome, but sudden blindness (the dread complication) is a catastrophe. Despite the conventional wisdom that the vertebrobasilar and internal carotid systems are not involved much, one group found that around 3% of biopsy-proven "temporal arteritis" patients had a stoke between the onset of symptoms and the initiation of glucocorticoid treatment (Medicine 88: 227, 2009). The glucocorticoids won't spoil the histopathology: J. Clin. Path. 65: 1138, 2012.
Many of these patients also suffer from pain and weakness in the muscles, the distinctive POLYMYALGIA RHEUMATICA (Lancet 381: 63, 2013). Future clinicians: The diagnosis is supported by finding everything normal on physical exam except perhaps for tender temporal arteries, plus normal labs except for a high "sed rate".
Only recently has it become clear that there is systemic overproduction of interleukins 1 and 6 for some reason, and activation of macrophages in the vascular intima. There is still a great deal that' s unknown (Ann. Int. Med. 139: 505, 2003).
Some folks will treat just on the clinical findings. Or prove your diagnosis with a temporal artery biopsy, which may or may not show granulomas on the inner elastic membrane. There is also a striking non-granulomatous fibrous proliferation of the intima. When in doubt, treat with prednisone or some similar glucocorticoid.
* On ultrasound, the aortic arch and its vessels are often thick as well (Rheumatology 51: 730, 2012).
* A histologic variant spares the intima but involves the vasa vasora / small vessels near the artery. Artifact of sampling, or separate entity, it's probably best to consider this temporal arteritis as well (Arth. Rheum. 64: 549, 2012).
* Methotrexate to supplement the prednisone for a more-effective, more-tolerable treatment: Arth. Rheum. 56: 2789, 2007.
* The British think the temporal artery biopsy should be 1 cm long (Br. J. Surg. 98: 1556, 2011).
* A variant involves the female genital tract (South. Med. J. 98: 469, 2005).
* How long to treat: Arch. Int. Med. 159: 577, 1999.
Polymyalgia rheumatica can occur in young people and/or in the absence of elevated sed rate (Arch. Int. Med. 157S: 317, 1997). When in doubt, treat.
* Future pathologists: Biopsies remain positive even after several weeks of glucocorticoid therapy (Br. J. Ophth. 86: 530, 2002).
{22095} temporal arteritis
{22096} temporal arteritis
{22098} temporal arteritis
{24777} temporal arteritis
{28019} temporal arteritis
TAKAYASU'S PULSELESS DISEASE ("aortic arch disease", etc.)
A fortunately-rare, idiopathic disease of younger adults (almost always) in which the aortic arch and its great branches thicken and their ostia become stenotic, strangling off blood flow to the upper part of the body.
No one knows the cause, and the histology is nonspecific, with granulomas, giant cells, lymphocytes, plasma cells, and so forth, in addition to the fibrosis and contraction.
* The molecular biology of temporal arteritis and Takayasu's is evidently similar. What's known: NEJM 349: 160, 2003. More on Takayasu's and temporal arteritis as a continuum: Medicine 88: 183 & 221, 2009. Watch for aspirin to be added to the glucocorticoid regimens, to prevent platelet-related intimal fibrosis in both diseases.
Whatever the real cause, surgical repair of the involved arch and branches is now giving excellent results (Ann. Thor. Surg. 81: 178, 2006). On medical therapy, or when surgery is not possible, the long-term prognosis is generally not good (Arth. Rheum. 56: 1000, 2007).
{48983} Takayasu's
COGAN'S DISEASE is another thankfully-rare disease usually affecting young adults. It features abrupt onset of nerve deafness, interstitial keratitis (you'll see it only on a slit lamp exam), and/or a systemic vasculitis often with aortic aneurysm formation. It's evidently caused by an autoantibody against inner ear and endothelium (Lancet 360: 915, 2002; update J. Ped. 160: 303, 2012).
KAWASAKI'S DISEASE ("mucocutaneous lymph node syndrome"; Ped. Clin. N.A. 46: 313, 1999; Am. Fam. Phys. 59: 3093, 1999; Lancet 364: 533, 2004; Heart 95: 787, 2009)
A febrile disease that resembles adult polyarteritis nodosa histologically but occurs in babies and toddlers, mostly of Japanese or Korean ancestry (no matter where they live now). The larger arteries get the most severe involvement.
The fact that almost all patients are around 2-5 years, the fact that occasionally an older child or adult gets the disease, the fact that there are outbreaks, and the fact that babies don't get it as long as they have maternal antibody all tell me the cause is an unidentified, ubiquitous virus.
* The established mouse model for coronary artery vasculitis / Kawasaki's involves immune activation by a bacterial extract: Circulation 125: 1542, 2012.
You'll want to see five of these six signs:
The most serious concern is coronary vasculitis, which causes myocardial infarcts. Healing can produce coronary aneurysms, etc. See Arch. Dis. Child. 87: 145, 2002.
* By electron microscopy, the endothelial cells are separated and perforated, rendering them hyperpermeable; again, no one knows why (Circulation 105: 766, 2002).
We treat Kawasaki's with aspirin and intravenous immunoglobulin. The outcome is good unless coronary disease becomes symptomatic.
Even in youngsters with coronary artery aneurysms, with today's interventions the long-term prognosis is pretty good (Circulation 123: 1836, 2011).
|
BUERGER'S DISEASE ("thromboangiitis obliterans": review
Angiology 47: 419, 1996; update Am. J. Med. Sci. 337: 285, 2009)
A disease of smokers, usually young men, in which the small neurovascular bundles in the extremities become inflamed and undergo thrombosis. No one has a clue as to the real etiology, beyond the link to tobacco smoking. It's also an ultra-rare complication of marijuana smoking (Br. J. Derm. 152: 166, 2005). The typical patient, after losing all his fingers, holds his cigaret between his last two toes. Not a pretty sight. The prognosis is hopeless unless the patient stops smoking. The anatomic pathology remains poorly worked-out. Neutrophils touching giant cells within thrombi is supposed to be characteristic. The most recent study (Virchows Archiv 436: 59, 2000) found instead that an intact internal elastic membrane, fibrosis much worse in the adventitia than anywhere else, endothelial swelling in the vasa vasora, and onionskinning of recanalization vessels were most helpful. |

|
INFLAMMATORY AORTIC ANEURYSM (JAMA 297: 395, 2007)
The overwhelming majority of patients are smokers. Otherwise, the etiology is unknown. I predict that the molecular cause of Buerger's and inflammatory aneurysm will be found at the same time.
Unlike classic atherosclerotic aneurysms, the outer surface is shiny-white with prominent little vessels, and usually there are fibrous adhesions to the nearby structures. The histopathology is intense inflammation and fibrosis of the adventitia.
INFECTIOUS ARTERITIS
Rickettsial disease, syphilis![]() ,
septic emboli (look for "Roth's spots" in the eyegrounds), walls of abscesses, and a host of
others.
,
septic emboli (look for "Roth's spots" in the eyegrounds), walls of abscesses, and a host of
others.
Worth mentioning here: A MYCOTIC ANEURYSM is a spot at a branch-point of an artery where a septic embolus (usually) has lodged and set up an infection, weakening the wall. ("Mycotic" is an unfortunate misnomer, since fungi aren't usually the culprits.)
Among the fungi, remember aspergillus and mucormycosis as especially good at invading vessels.
RAYNAUD'S DISEASE / PHENOMENON
Spasm and occlusion of the arteries supplying the fingers, which turn white ("pallor"), then red ("suffusion"), then blue ("cyanosis"). Triggered by cold weather, it's most often idiopathic; known causes range from vasculitis syndromes to operating jack-hammers.
Scleroderma patients and some others have this process greatly exacerbated by hyperplastic arteriolar sclerosis in the digital arteries.
If it's a bother, get out the calcium-channel blockers, and/or a nice warm pair of gloves.
{24503} Raynaud's
{25459} Raynaud's
{39657} Raynaud's
{39654} Kawasaki's?
{39655} Kawasaki's?
{48983} Takayasu's
The common pathophysiology is loss of vascular control; they will first become constricted or blocked, then dilate and become inflamed.
The red color of affected body parts probably results from opening of arteriovenous channels and closure of precapillary sphincters. Lack of nutritive blood flow contributes to the pain, while the excess non-nutrative blood flow causes a burning sensation.
Long-mysterious, it's now pretty clear that it's a often a small-fiber neuropathy (Arch. Derm. 139: 1337, 2003; Brain 126: 567, 2003; update Anes. Anal. 104: 438, 2007).
There's a hereditary form with mutated neuronal sodium channels (Neurology 67: 1538, 2006), and it is possible that the adult acquired form results from autoantibodies against this channel (Am. J. Med. Sci. 335: 320, 2008). These patients present a major pain-management problem -- Mayo's has a series (Arch. Derm. 144: 1578, 2009).
Myeloproliferative diseases with extremely high platelet counts also produce erythromelalgia, probably by the platelets plugging vessels.
ATHEROSCLEROTIC AORTIC ANEURYSMS
|
|
|
ATHEROSCLEROSIS often causes aneurysms, usually distally in the aorta, above the level of the iliacs. When the abdominal aortic diameter exceeds 3.0 cm, it's an aneurysm. Exceed 5 cm or so and it's likely to burst (retroperitoneally, intra-peritoneally, intra-duodenal), with (usually) lethal consequences. Patients may complain first of back pain, etc. Aneurysms are always lined by thrombus (why?), which sometimes embolizes. Iliac aneurysms are also common; basilar artery aneurysms seldom rupture but may compress important things.
Occasionally these get infected; the usual bug is salmonella (Am. J. For. Med. Path. 23: 382, 2002).
* Albert Einstein's physicians knew he had an aneurysm, but when it burst, they got focused on his gallbladder instead. He died as a result.
{03665} atherosclerotic aortic aneurysm
{11042} atherosclerotic aortic aneurysm
{11048} atherosclerotic aortic aneurysm, repaired
{11642} atherosclerotic aortic aneurysm
{11645} atherosclerotic aortic aneurysm
{18717} atherosclerotic aortic aneurysm
{20305} atherosclerotic aortic aneurysm
{24780} atherosclerotic aortic aneurysm
{25742} atherosclerotic aortic aneurysm
{04589} atherosclerotic basilar artery aneurysm
{24836} atherosclerotic aneurysm, brain
|
|
![]() Coronary Artery Aneurysm
Coronary Artery Aneurysm
Classic drawing
Adami & McCrae, 1914
SYPHILIS![]() ("lues")
causes ischemic damage (by narrowing / occluding vasa vasora) to the walls of the arteries, and is
famous for causing proximal aneurysms that rupture impressively. Before rupture occurs, look
for the infamous "tree bark" grooves on the intima, as well as occlusion of the coronary and other
ostia and compromise of the aortic valve ring. It's still very much around
(Ann. Thorac. Surg. 92: 1503, 2011).
("lues")
causes ischemic damage (by narrowing / occluding vasa vasora) to the walls of the arteries, and is
famous for causing proximal aneurysms that rupture impressively. Before rupture occurs, look
for the infamous "tree bark" grooves on the intima, as well as occlusion of the coronary and other
ostia and compromise of the aortic valve ring. It's still very much around
(Ann. Thorac. Surg. 92: 1503, 2011).
Historically, we've taught that proximal aortic aneurysms are usually due to syphilis ("luetic"). Nowadays, syphilis is very rare, and atherosclerosis is common. I wouldn't assume an ascending aortic aneurysm is luetic unless the arch is free of significant atherosclerosis.
* "Tree-barking" is just stretch-marks of the aorta. The only reason this is supposed to be "more typical of syphilis than atherosclerosis" is that, in atherosclerosis, the intima is already distorted by the atherosclerotic plaques. I know this, because in my several cases of Marfan-style dilatation of the aortic root, there's always been tree-barking.
{10224} syphilitic aneurysm
{18716} syphilitic aneurysm
![]() Syphilitic aneurysm
Syphilitic aneurysm
Classic patient photo
Adami & McCrae, 1914
|
|
AORTIC DISSECTION
DISSECTING HEMATOMA, often miscalled "dissecting aneurysm", is blood that has entered the wall of the aorta and is following a weak plane ("cystic medial necrosis", actually there are no cysts and no necrosis, just diminished elastic fibers and maybe a little extra mucoid goo).
Think of the blood acting as a chisel under the strokes of the heart.
* A study that questioned whether the elastic is extensively damaged in most people who have experienced acute ascending aortic dissection (Am. J. Card. 108: 1639, 2011; all patients were or had been hypertensive) doesn't square with your lecturer's small series; wait and see on this one.
The lesion often happens over days or longer, and is infamously missed by physicians who didn't listen for aortic regurgitation. People coming to autopsy after death from thoracic aortic dissections have often (1 in 3) visited their physician during the previous week, and generally have very large hearts (from hypertension and/or from aortic regurgitation) -- don't miss it (Am. Heart. J. 162: 474, 2011).
![]() Aortic dissection
Aortic dissection
Classic drawing
Adami & McCrae, 1914
![]() Aortic dissection
Aortic dissection
Dr. Heuser
Wikimedia Commons
![]() "Cystic medial necrosis"
"Cystic medial necrosis"
WebPath Photo
This catastrophe results in progressive compromise of arteries, backwards rupture damaging the aortic valve and/or coronary ostia, or further backwards rupture into the pericardial sac or pleural space.
Future clinicians: "Stanford type A" lesions begin at the aortic root or ascending aorta and are treated surgically; without surgery, most (not all Ann. Thor. Surg. 96: 1066, 2013) patients die. "Stanford type B" lesions begin in the arch or descending aorta.
Patients experience a "ripping", agonizing chest pain as the false lumen expands. Michael DeBakey's claim to fame is having classified and devised the surgical treatment for these people. Otherwise, the patient's best hope is to have the blood re-enter the lumen, establishing a "double-barrel aorta", with the false lumen eventually becoming covered with normal endothelium. Or the process may simply stop.
Marfan types are more prone to this lesion, but nobody's immune. An epidemic of dissecting aneurysms occurred among turkeys who acquired osteolathyrism after eating beta-NH2-propionitrile in sweet peas.
* Having a congenitally bicuspid aortic valve seems to increase the risk of an aortic dissection starting near the valve considerably: Heart 99: 1668, 2013.
* The most spectacular multiple aneurysms in medicine are perhaps seen in the Marfan-like Loeys-Dietz syndrome, caused by a mutation in one or the other receptor for transforming growth factor beta (Nat. Genet. 37: 275, 2005).
* Nowadays, if one of your parents or siblings had an aortic dissection, you go get your genes checked; at this writing (2013), seven loci for "familial thoracic aortic aneurysm and dissection" are known.
Minor variants, with limited extension of a bleed into the aortic wall or down around the root, also exist. These may or may not turn into the fully-expressed, deadly disease.
Trauma to the aorta (for example, a car wreck) or another major artery (infamously, the vertebral arteries in a very bad neck manipulation) can cause dissection of blood down a torn media.
"Spontaneous" dissection of arteries in the neck can cause stroke in young people (NEJM 330: 393, 1994).
Aortic dissection resulting from a catheter mishap is well-known, with a risk of about 0.7% (Am. Heart J. 159: 1147, 2010).
* Non-surgical management using a stent-graft: NEJM 340: 1585, 1999 (wow!).
{17467} dissecting aneurysm
{18718} dissecting aneurysm
{20222} dissecting aneurysm
{25747} dissecting aneurysm
|
|
CORONARY ARTERY DISSECTION is thankfully rare. For some reason, it most commonly occurs after childbirth.
VEINS
VARICOSE VEINS
A self-perpetuating process caused by loss of competency in the leg vein valves and support structures.
We lose our elastic fibers as a result of aging, or blow out valves (by getting fat, having babies, straining at stool, standing up doing surgery). The tendency to get varcose veins is hereditary.
The weight of the column of blood doesn't make life easier for the next valve or the support of the next few cm of vein. Get elastic leg wraps and hope for the best. If it gets too hard to pump blood back to the heart by way of the veins, or the weight of the column of blood causes continuous micro-hemorrhages, you will see the familiar hemosiderin STASIS PIGMENTATION and/or ischemic STASIS ULCERS.
Stasis ulcers themselves are a very common, very hard-to-treat problem of older folks. Older explanations ("the poor circulation produces hypoxia") are being supplemented by new work (the weight of blood causes capillaries to rupture, with red cells and plasma proteins being extravasated, with local cytokine activation; Surg. Clin. N.A. 83: 671, 2003.) This fits with there usually being pigmentation here, and with the accompanying local fibrosis (lipofibrosclerosis).
![]() You already know from physiology about ESOPHAGEAL VARICES and
HEMORRHOIDS as evidence of portal
hypertension.
You already know from physiology about ESOPHAGEAL VARICES and
HEMORRHOIDS as evidence of portal
hypertension.
Fortunately, thrombi that may form in varicose veins very seldom embolize.
THROMBOPHLEBITIS ("trombone fleabites", better "phlebothrombosis"; reviews Lancet 353: 479 & 1167, 1999)
Thrombosis of a deep vein, most often in the leg. You're already familiar with the causes, and the dread complication (pulmonary embolus). Usually, pain and swelling result, hence "-itis".
Vicious variants include MILK LEG ("phlegmasia alba / cerulea dolens", thrombosis of the iliac vein, around the time of parturition), BUDD-CHIARI SYNDROME (thrombosis of the hepatic veins) and DURAL SINUS THROMBOSIS.
TROUSSEAU'S MIGRATORY THROMBOPHLEBITIS affects first one vein, then another. The cause is usually cancer of the pancreas, less often another adenocarcinoma.
SUPERIOR VENA CAVA SYNDROME
From occlusion, usually from extrinsic compression by lung cancer.
Patients have dusky skin in the head, upper extremities, and upper chest.
"SVC syndrome" can cause a bad headache just from the markedly increased venous pressure.
INFERIOR VENA CAVA SYNDROME
From occlusion, usually by cancer in para-aortic lymph nodes, or a thrombus-plugged IVC filtre.
Patients have dusky lower bodies and unusual patterns of collateral circulation.
LYMPHATICS
LYMPHANGITIS
Inflammation of lymphatics, usually by a bacterium that's good at invading them (i.e.,
streptococcus![]() group A.) You'll see red streaks running along an extremity, etc.
group A.) You'll see red streaks running along an extremity, etc.
LYMPHEDEMA
Compromise of lymphatic draining of the extremities and/or genitalia. The most important causes are cancer of the cervix, filariasis, iatrogenic (i.e., after the overly-zealous old mastectomies), autosomal dominant ("Milroy's disease", the lymphatics don't form properly), or "idiopathic" (again, sometimes the lymphatics don't form properly). Turner's girls often have lymphedema of the hands and feet.
If there's hyperplasia of the overlying epidermis, it's ELEPHANTIASIS.
"Massive localized lymphedema" looks clinically and microscopically like a sarcoma, but results from lymphatics obstructed by masive obesity (J. Clin. Path. 62: 808, 2009).
![]() Lymphedema
Lymphedema
Patient education site
By a cyberfriend
TUMORS OF BLOOD VESSELS
The cell of origin is the endothelial cell. When the vascular origin of a tumor isn't obvious, we confirm the diagnosis using immunostains (von Willebrand's factor, CD31, CD34); old-timers found Weibel-Palade bodies in the endothelium on electron microscopy.
HEMANGIOMAS
Mostly hamartomas. You'll see a variety of these clinically.
* Future pathologists: There are some rare, exotic tumors of blood vessel origin. Don't worry about them.
CAPILLARY HEMANGIOMAS (little vessels) and CAVERNOUS HEMANGIOMAS (big vessels "resembling erectile tissue") are common on the skin. STORK BITES (backs of the neck and/or forehead of a baby) usually involute (i.e., thrombose and organize) after a few years, while CHERRY ANGIOMAS of the skin start popping up after age 20 or so.
{05888} hemangioma
{12235} hemangioma
{13041} hemangioma
{13042} hemangioma
{13044} hemangioma
{13045} hemangioma
{13047} hemangioma
{13048} hemangioma
{13050} hemangioma
{13051} hemangioma
{13141} hemangioma
{17507} hemangioma in liver
{21832} hemangioma
{21834} hemangioma
{21835} hemangioma
{22116} hemangioma
{22118} hemangioma
{22119} hemangioma
|
|
PORT-WINE STAIN (one form of "nevus flammeus"), fashionable in the Gorbachev era, can be treated by laser if the patient wishes. A hemangioma in the meninges (bad, shunts blood away), generally with an overlying port-wine stain in the V1 area (or check the eyelids; Pediatrics 87: 323, 1991), is STURGE-WEBER syndrome (somatic mutation in GNAQ NEJM 368: 1971, 2013).
{53705} Sturge-Weber
PYOGENIC GRANULOMA (noted misnomer; neither pyogenic nor a granuloma) pops up the gums of pregnant ladies, or the skin of anybody. Grossly, it looks like a rotten cherry and bleeds very easily. Microscopically, it looks like granulation tissue.
{13141} "pyogenic granuloma"
{12786} "pyogenic granuloma"
{12789} "pyogenic granuloma"
{12225} pyogenic granuloma
* EPITHELIOID HEMANGIOMAS are curious bumps of small vessels surrounding a single large one; the connective tissue between the vessels is packed with eosinophils.
* PAPILLARY ENDOTHELIAL HYPERPLASIA is just an unusual way way for a thrombus to organize. Endothelial cells make little papillae. The lack of anaplasia distinguishes it from an angiosarcoma.
Large hemangiomas can consume a person's platelets, the serious KASABACH-MERRITT SYNDROME (Am. J. Clin. Path. 99: 628, 1993; a kid received 6622 units of platelets over 19 months, a new record; Am. J. Med. Sci. 333: 293, 2007 disseminated angiosarcoma).
{12237} Kasabach-Merritt giant hemangioma
KLIPPEL-TRENAUNAY SYNDROME is a huge capillary hemangioma plus enlargement-deformity, always from a somatic mutation (* gene VG5Q: Nature 427: 640, 2004). A single extremity (or perhaps more than one) is involved. Mayo Clin. Proc. 73: 28, 1998.
| * Golfer Casey Martin is the best-known Klippel-Trenaunay patient. In 2001, the US Supreme Court ruled (PGA tours vs. Martin) he could ride in a cart despite the PGA's complaining it would give him an unfair advantage. |
VON HIPPEL-LINDAU anti-oncogene deletion syndrome results from mutated VHL. It features the most troublesome hemangiomas in all of medicine, as well as renal cell carcinoma.
GLOMANGIOMAS (glomus tumors)
Painful tumors of the smooth muscle of the vestigial human GLOMUS organs, little thermoregulatory left-overs from mammalian evolution and found in the fingertips, toes, and coccyx (the rat's tail is the most familiar glomus).
{09017} glomus tumor
TELANGIECTASIS
"Dilatations of the ends of little vessels".
OSLER-WEBER-RENDU TELANGIECTASIAS ("hereditary hemorrhage telangiectasia") result from any of several autosomal dominant genes (* some, but not all, patients lack a binder for transforming growth factor beta). Most do not know they are affected, and simply they think they have "frequent nosebleeds." Patients have little vascular malformations connecting little arteries and little veins along their whole GI tract (often easiest to see on the lips, of course), and often lots of other places. Especially, AV malformations in the lungs (where they tend to be large) can produce right-to-left shunts, which are dangerous. The lesions in the nose and GI tract are prone to bleed. Reviews NEJM 333: 918, 1995; NEJM 345: 325, 2001; clinicians Am. Fam. Phys. 82: 785, 2010.
* Many of these patients also have "idiopathic pulmonary hypertension", also from faulty vascular development; depends on the allele).
The familiar "liver spider" is a centrally dilated artery supplying several little arterioles that blanch when the "spider's body" is pressed.
CANCERS OF THE BLOOD VESSELS
HEMANGIOENDOTHELIOMAS are low-grade cancers of endothelium. They makes little vessels. Leave the diagnosis to the pathologists.
(HEM)ANGIOSARCOMAS (Arch. Path. Lab. Med. 133: 1804, 2009; Ann. Surg. 251: 1098, 2010): A host of aggressive cancers. Best-known was epidemic hepatic angiosarcoma, caused by exposure to vinyl chloride or "Thorotrast" contrast medium. Hopefully that's rare today, but occcasionally sun-exposed skin develops angiosarcomas. At other sites, they often follow therapeutic radiation. Update Arch. Path. Lab. Med. 133: 1804, 2009.
Angiosarcomas are treacherous, especially to pathologists, and often look deceptively benign. There may be little or no anaplasia. Any "hemangioma" that isn't nicely circumscribed should raise the suspicion of angiosarcoma.
* Don't worry about the hated "atypical vascular lesion", the super-low-grade angiosarcoma that arises in the papillary dermis, after radiation or by itself.
{10863} hemangiosarcoma
{21065} angiosarcoma, breast
{40635} angiosarcoma
HEMANGIOPERICYTOMAS: Low-grade cancer of the pericytes. As you'd expect, the tumor cells interlace with vessels, beautifully demonstrated in reticulin-stained preparations. Again, leave the diagnosis to the pathologists.
{09484}* hemangiopericytoma. This is subtle.
{09486}* hemangiopericytoma
{09489} hemangiopericytoma, reticulin stain
{09490} hemangiopericytoma, reticulin stain
KAPOSI'S "SARCOMA" (Arch. Path. Lab. Med. 137: 289, 2013): The business cell is endothelium, and it can spread and choke off the viscera.
The cause, of course, is herpes virus 8![]() in people who are somewhat immunocompromised; the more severe the immune disability, the more severe the Kaposi's.
in people who are somewhat immunocompromised; the more severe the immune disability, the more severe the Kaposi's.
Classic American Kaposi's of the pre-AIDS era mostly involved the legs of older men, and seldom caused major problems.
Renal transplant patients are prone to yet another Kaposi's variant.
LYMPHANGIOMAS: Hamartomas. The best-known is "cystic hygroma" of the necks of babies.
{23443} lymphangioma
{13392} cystic hygroma
|
|
|
|
|
LYMPHANGIOSARCOMAS: Cancers of the lymphatics, generally arising in lymphedema (i.e., after mastectomy, called * "Stewart-Treves syndrome") or after exposure to radiation.
HIGH TECHNOLOGY
ANGIOPLASTY is surely familiar to you.
Further, the lesion will heal by proliferation of the local cells, and it's about an even chance that in six months, the artery will be just as narrow.
What has helped most in preventing these problems is the placing of metal stents.
DEATHS DUE TO BEVACIZUMAB TREATMENT OF CANCER (JAMA 305: 487 & 506, 2011). Bevacizumab "Avastin" is the fantastically-expensive humanizied antibody against VEGF, which causes blood vessels to form in tumors and normal healing injuries / granulation tissue. It seems to give some cancer patients a few extra good months (outcomes of studies are very mixed; one massive, expensive, total failure in advanced lung cancer is documented in Lancet 377: 1846, 2011), but not surprisingly recipients tend to die of hemorrhages whether or not it slows their cancers. (Neutropenia and bowel performation are also linked -- why might this be?) If we are to believe the JAMA writers, one has perhaps 1 chance in 40 that bevacizumab being added to your chemotherapy will kill you.
* After thirty years of "study" in the U.S., evidence that acupuncture actually works for ANY disease of the cardiovascular system remains conspicuous by its absence. The best available is that some hypertensives' blood pressure drops some while they are actually in the mellow, comforting acupuncturist's office, only to return to their usual levels when the treatments are over (Circulation 103: 2038, 2001). A major recent study at and near Harvard (SHARP -- Stop Hypertension with the Acupuncture Research Program) has been undertaken, using sham acupuncture for controls (Cont. Clin. Tri. 25: 76, 2004); I predicted in 2004 that it would not publish positive results, and judging on the data (Hypertension 48: 838, 2006) it was a total failure in every category. It's to the great credit of the New England School of Acupuncture, which did a lot of the work, that they were up-front about the whole thing.
* "How many miles of blood vessels are there in a pound of fat?" People write me about this every once in a while. Let's figure it out. Assume an adipocyte is 50 microns across; it'll vary from 10-100 depending on how fat the person is. The fatter you are, the less vascular is your fat, which is one more reason that this whole inquiry is silly. In a section of body fat, which I examine often enough under the microscope, the capillary (there has to be at least one) that supplies each fat cell is not usually visible, so I'll assume one per adipocyte, and all going in the same direction. Put a single capillary between each pair of fat cells and that's about 20 capillaries per millimeter, or about 500 capillaries per inch, or 250,000 capillaries per square inch. Assume a pound of fat is a cube 4" on a side, which is good enough for junk science, or 16 square inches, and that is 4,000,000 capillaries running through the cube, 16,000,000 inches. There are 12 inches in a foot and 5280 feet in a mile, so if you get 500 miles you did the arithmetic the same way that I did. If you prefer 100 miles as in other estimates, simply assume that there's a capillary between every other pair of adipocytes, rather than every pair. That this question is fundamentally wrong-headed can be understood by anyone who considers whether moving a certain total number of cars through Kansas City would be easier with more highways or fewer highways. Further, the vast majority of these capillaries are completely closed at any moment during your life, and not carrying any blood. At autopsy, blood usually dribbles from other organs but not from fat. At surgery, other organs bleed plenty but fat barely bleeds. The real question isn't, "How many extra miles of blood vessels?", but "How much rougher is it on my heart to be fat?" Think about walking around carrying 100 lb of weights everywhere you go. The truth is that "education" and moral exhortation do not cause people to lose weight; overeating is programmed just like scratching when you itch.
 * SLICE OF LIFE REVIEW: VESSELS AND HEART
* SLICE OF LIFE REVIEW: VESSELS AND HEART
{03184} tricuspid valve, normal
{03187} tricuspid valve, normal
{03329} atrium, normal
{03344} hypertrophy & normal, (l) vent.
{03347} hypertrophy & normal, (l) vent.
{03401} comparison of atria, normal ra & la free walls
{03467} heart, normal histology
{03575} ventricular slice method of dissection normal heart
{03581} cardiac base method of dissection, normal heart
{03599} short axis dissection, normal heart
{03605} four chamber dissection, normal heart
{03611} long axis dissection, normal heart
{03629} cardiomyopathy, dilated; normal
{03683} pericardium intact opened, normal
{03686} pericardium opened removed, normal
{03689} pericardial sac, normal with and without heart removed
{03692} pericardium, normal with anterior portion removed
{03698} heart anterior, normal
{03716} heart, normal (r) (l) ant. oblique
{03719} heart anterior, normal
{03734} heart, normal (r)
{03749} heart, normal
{03758} tricuspid valve, normal
{03764} tricuspid valve (r) ventricle view, normal
{03767} mitral valve opened, normal
{03776} mitral chordae, normal
{03779} mitral valve, normal viewed from LA and LV aspects
{03785} aortic-mitral valvular continuity, normal
{03788} tricuspid mitral valves, normal comparison of shapes
{03791} tricuspid mitral valves, normal
{03794} heart, normal
{03797} heart, normal right ventricle
{03800} heart, normal left ventricle
{03803} heart, normal compare ventricle thick.
{03806} heart, normal
{03809} heart, normal
{03812} heart, normal

{03815} heart, normal: how to cut
{03818} heart, normal: how to cut
{03821} heart, normal: how to cut
{03851} myocardial cell, normal left ventricle
{03854} membranous septum (l) ventricle, normal
{03857} membranous septum (r) ventricle, normal
{03863} pulmonary valve, normal
{03866} aortic valve, normal
{03869} aortic valve closed opened, normal
{03872} aortic valve cusp in elderly, normal
{03881} aortic valve from above, normal
{03884} cardiac valves (all four), normal
{03899} coronary ostia, normal
{03902} aortic valve coronary ostia, normal
{03905} aortic valve coronary ostia, normal
{03908} coronary arteries, normal
{03911} coronary arteries sup. inf., normal
{03914} coronary artery dominance, normal
{03920} septum perforators of (l) ant. desc., normal
{03923} septum coronary arteries, normal
{03926} coronary arteries, normal
{03929} coronary arteries, normal
{04472} hear, normal
{04475} aorta, normal
{06218} superior vena cava aorta pulm. artery, normal
{06224} aortic arch, normal longitudinal section
{06233} vein, normal
{06539} coronary angiogram, normal
{09464} artery, normal
{09828} artery, normal
{11036} artery, normal
{11402} vertebral basilar artery, normal
{11405} vertebral basilar artery, normal
{11408} vertebral basilar artery, normal
{11738} heart, normal ventricle myocardium
{11799} heart, normal
{11800} heart, normal
{11809} aorta, intimal surface
{12889} venogram, normal
{12892} venogram, abnormal
{12894} venogram, abnormal
{14627} Purkinje fibers cross section
{14628} Purkinje fibers long.
{14687} capillary, normal
{14689} arteriole-venule, normal
{14692} artery, muscular
{14693} artery, muscular; shows internal elastic membrane
{14694} artery, small muscular
{14695} artery, small muscular
{14696} artery, muscular
{14697} artery, muscular; b=media, c=external elastic, d=adventitia, e=nerve
{14698} artery, vein
{14699} artery, vein
{14700} aorta, normal
{14701} aorta, normal (elastic stain)
{15047} adrenal medulla (central vein), normal
{15180} stomach, cardia
{15181} stomach, cardia
{15209} Purkinje fibers, heart
{15210} epicardium, #17
{15211} endocardium, #17
{15212} epicardium, #17
{15213} endocardium, #17
{15214} atrium, endocardium
{15215} atrium, endocardium
{15217} artery and vein, elastic stain
{15218} artery, #29
{15219} vein, #29
{15220} aorta, #22, elastic stain
{17466} artery, normal
{20234} ecg, normal
{20255} chest, normal x-ray
{20567} insufficiency, aortic with normal on r
{20798} vein
{20816} Purkinje fiber, heart
{20817} myocardium in cross section
{20818} epicardium with fat and blood vessel
{20819} myocardium with epicardium too
{20820} endocardium
{20821} atrium of heart, endocardial surface
{20822} atrium of heart, endocardial surface
{20823} atrium of heart, epicardial surface
{20824} artery and vein, longitudinal section
{20825} artery and vein, cross section
{20826} artery, elastic
{20839} artery
{31412} internal carotid artery, normal
{31442} straight sinus nerve vein of Galen, normal venous anatomy
{37643} heart, normal
{38015} artery, normal
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about heart pathology to consult these standard textbooks.
In my notes, the most helpful current journal references are embedded in the text. Students using these during lecture strongly prefer this. And because the site is constantly being updated, numbered endnotes would be unmanageable. What's available online, and for whom, is always changing. Most public libraries will be happy to help you get an article that you need. Good luck on your own searches, and again, if there is any way in which I can help you, please contact me at scalpel_blade@yahoo.com. No texting or chat messages, please. Ordinary e-mails are welcome. Health and friendship!


| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |

 Blood Vessels and Heart
Blood Vessels and Heart
 Benign lymphangioendothelioma
Benign lymphangioendothelioma