Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
verify here. |
Cyberfriends: The help you're looking for is probably here.
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- Heart
Lectures follow Textbook
QUIZBANK
Heart (all)

{08187} Heart within the pericardium
What's a 'double-blind study'? Two pathologists trying to read an EKG!
A simple rule to follow, whatever you do: If you're not having fun, you're not in the right place.
--Anonymous (and of course baseless)
-- Joe Montana, "Art and Magic of Quarterbacking"
Once I had brains, and a heart also; so having tried them both, I should much rather have a heart.
--The Tin Woodsman of Oz
Does CPR work better if you do your compressions with a toilet plunger? The great controversy, including a frank admission that CPR....: JAMA 273: 1299, 1995. Twenty years later: CPR outcomes are still dismal and the only advantage of the CPR Machine seems to be that it allows ambulance personnel to wear their seat belts: Lancet 385: 947, 2015. When North Carolina undertook a huge public health initiative to teach everybody CPR ("bystander"), survivals rose to 33% but there was "favorable neurological outcome" in only 9% (JAMA 314: 255, 2015). How many of those with a good outcome really needed CPR isn't clear from the paper and probably can't be determined. Similar results from Japan (JAMA 314: 247, 2015). Is that really "good news"?
A talking monitor-defibrillator now exists that tells resuscitation teams how well they are doing and how well they are complying with guidelines. It clearly helps teams do things right, and after 1586 uses had no demonstrable impact at all on any outcome measure (BMJ 342: d512, 2011).
Out-of-hospital CPR survivors do twice as well (i.e., after you exclude those that were already obviously hopeless, maybe 6% of them are alive without obvious brain damage a month later) if you DON'T do the mouth-to-mouth stuff...: Lancet 369: 920, 2007.
Your instructor's personal decision not to allow CPR to be performed on him is not to be taken as rejection of the work that has gone into developing or practicing the technique, or a denial that some folks are saved by it and leave good quality lives afterwards. We can chat about this if you like.
According to the British, the United States of America now spends 0.5-1% of our ENTIRE gross national product on the critical care unit, with the vast majority of the patients not living long (Lancet 376: 1273, 2010).
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

Define and use the following terms:
Describe the changes in the myocardium in a trained aerobic athlete, and recognize that these are desirable rather than harmful.
Review the general pathology of congestive heart failure. You should probably already know this from your earlier studies of cardiac physiology and the pathology of the body fluids.
Describe the clinical spectrum of atherosclerotic coronary artery disease.
Tell how the various kinds of angina pectoris arise. Explain how myocardial infarcts occur, why they are so serious, and what the pathologist will see at autopsy under varying circumstances. Tell how subendocardial infarcts occur. Describe the typical picture of chronic ischemic cardiac disease. Tell what a pathologist will find in classic coronary "sudden death", and when the diagnosis can and cannot be made.
Mention the other causes of ischemic heart disease, and tell about how they operate. Tell about the other causes of sudden death.
|
Define and use the following terms:
atresia
|
Alan Shepard |
Recall the upper limit of normal weight of the sedentary adult's heart, left ventricular thickness, and right ventricular thickness.
List the minimal anatomic criteria for hypertensive heart disease. Describe the role of pressure overload (clear) and chronic catecholamine stimulation (probable) as causes of this hypertrophy. Describe the gross and microscopic changes typical of the hypertensive's heart. Appreciate that this is a very common finding, both clinically and at autopsy. Explain the difficulty of making the diagnosis when left ventricular failure confuses the picture.
Distinguish cor pulmonale from right heart enlargement caused by left ventricular failure or by congenital malformations. Recognize pulmonary embolization as the only setting for true "acute cor pulmonale". Describe the hypoxic vascular response, and describe how the shape of the right ventricle on cross section differs from normal in this setting. Recognize cor pulmonale as a sufficient explanation for sudden death cases coming to autopsy. Appreciate the tremendous clinical importance of cor pulmonale in many settings.
Tell what we know about causes of serious congenital heart disease. Recall the incidence per thousand live births, and the risk of recurrence. List the problems common to all these children, and the hazards presented by jet lesions.
List tetralogy of Fallot (most common), transposition of the great arteries, persistent truncus arteriosus, and tricuspid atresia as the four most important forms of congenital cyanotic heart disease, and clearly explain the abnormal anatomy and physiology of each. Explain the seriousness of the right-to-left shunt, the most dreaded consequences of paradoxical embolization, and other problems faced by these patients.
Diagram tetralogy of Fallot, listing the four features that define the syndrome.
Describe the usual pattern in transposition of the great arteries, and how a septal defect permits survival after birth. Distinguish "corrected transposition".
Define truncus arteriosus, and recall that it leads eventually to pulmonary hypertension.
Recall ventricular septal defect, atrial septal defect, and patent ductus arteriosus as the major causes of congenital left-to-right shunt. Explain the associated hazards, and especially why cyanosis develops weeks to years after birth in patients with left-to-right shunts.
Recall the location of most ventricular defects. Explain the reason that a "VSD" is unwholesome. Give the meaning of "Roger's disease" and the spontaneous closure rate.
Describe the usual clinical course in atrial septal defect, and tell why these are so seldom recognized in youth. Distinguish ostium primum ("endocardial cushion"), ostium secundum, and sinus venosus atrial septal defects. Recognize ostium primum as the usual form in Down's syndrome, as ostium secundum as most common in other people.
Locate the normal ductus arteriosus and define its function and fate. Identify prostaglandin E as maintaining the patency of the normal ductus. Recognize that in congenital heart disease with impaired blood flow to the lungs, it is good for the ductus to remain open.
Describe patent ductus arteriosus, mentioning its relationship to other defects and to Turner's syndrome, and its most common location. Tell why preductal coarctation can cause right sided heart failure in utero. Describe how it causes hypertension, and mention clinical findings that would alert the pediatrician to post-ductal coarctation. Mention the reasons for getting it fixed surgically.
Recognize pulmonary stenosis with intact interventricular septum as a common, serious cardiac malformation.
Describe the problems caused by a bicuspid aortic valve. Describe the aortic valve in congenital valvular aortic stenosis, and the defect in congenital sub-valvular stenosis. Explain why aortic stenosis commonly produces sudden death.
Give a short account of each:
Anitschkow cell / Aschoff body
antihyaluronidase
antistreptolysin O ("ASO")
Barlow's syndrome / degenerative mitral valve disease
caterpillar cell
dextrocardia with situs inversus
dextrocardia, isolated
erythema marginatum
friable
Kartagener's syndrome
lines of closure
MacCallum's patches of the left atrium
mid-systolic click
regurgitation
Roth spots
situs inversus totalis
splinter hemorrhages
Sydenham's chorea ("St. Vitus Dance")
tamponade
valvular insufficiency
valvular stenosis
vegetations
Relate dextrocardia, Kartagener's syndrome, immotile cilia, and situs inversus totalis.
Remember that mitral stenosis is virtually always caused by scarring from rheumatic fever.
List the important causes all eight valvular syndromes.
List the three important causes of acquired aortic valve stenosis. Sketch a normal (three cusp) aortic valve with calcific stenosis, mention the age of onset, and explain why the process is so serious. Sketch a bicuspid valve with the same thing, and mention which kind of valve is more prone to this.
Describe Barlow's "syndrome" of the mitral valve. Tell how prevalent the disease is, describe the relationship to Marfan's syndrome, and tell what makes the mid-systolic "click". Describe the four complications (bacterial endocarditis, mitral insufficiency, rhythm disturbances, and cardiac neurosis) that can result.
Describe the essential pathogenesis of rheumatic fever and rheumatic pancarditis, and the typical time of onset. List the six principal findings, and describe the changing incidence of the disease in the U.S. and globally. Mention the recurrence rates cited after repeat strep throat. Describe a typical Aschoff body, and tell where it is located. Explain why rheumatic endocarditis is considered more serious in the long run than the myocarditis or pericarditis, and describe the locations of the lesions on the valves in the acute illness.
Describe the pathologic anatomy in chronic mitral valve deformity and chronic aortic valve deformity following rheumatic fever.
Explain why infective (i.e., bacterial) endocarditis is so serious. Tell ways in which the blood becomes seeded with microbes, and times and places where the fibrin-platelet thrombi form inside the heart. Describe acute infective endocarditis, the types of valves that may be involved, its usual cause, and the fatality rate. Name the bacterium most often responsible for subacute bacterial endocarditis. Tell what you will see grossly and microscopically. Tell what valve are most often involved in IV drug-users and in other people. Mention why bacterial endocarditis might be "culture negative". Describe the dread complications of bacterial endocarditis in some detail, and mention clues to the diagnosis. Tell what healed bacterial endocarditis looks like.
Describe typical settings for nonbacterial thrombotic endocarditis ("marantic endocarditis"). Describe the gross and microscopic lesions.
Describe calcification of the mitral annulus as seen in some older individuals, and describe its clinical significance.
Describe the gross, microscopic, and functional lesions in carcinoid syndrome, and explain why we think that the lesions usually occur only on the right side.
Recognize the five complications of valve replacement.
Give a short account of each:
adriamycin
cardiomyopathy
Chagas's disease
daunorubicin
doxorubicin
effusion
myocarditis
Distinguish "myocarditis" (i.e., inflammatory, i.e., autoimmune or infection) and "cardiomyopathy" (i.e., a noninflammatory disorder).
Describe the gross and microscopic pathology and clinical course of a typical case of myocarditis.
Recall viruses, especially Coxsackie A![]() & B
& B![]() and parvovirus B19 as the most important causes of significant acute
myocarditis, and that this is (fortunately) rare. Mention why we think much of the damage is
immune-mediated. Explain why we think many cases of "idiopathic dilated cardiomyopathy"
("Barney Clark's disease") result from Coxsackie myocarditis.
and parvovirus B19 as the most important causes of significant acute
myocarditis, and that this is (fortunately) rare. Mention why we think much of the damage is
immune-mediated. Explain why we think many cases of "idiopathic dilated cardiomyopathy"
("Barney Clark's disease") result from Coxsackie myocarditis.
Given a cardiomyopathy, subclassify it as dilated ("flabby heart"), hypertrophic ("muscle-bound heart"), or restrictive-infiltrative-obliterative (i.e., amyloid, "stiff heart").
Recall "dilated-congestive" cardiomyopathy as an end-stage of various longstanding cardiac injuries, and describe the way this heart looks and functions. Describe the histology, and why mural thrombi form.
Cite the clinical features of alcoholic cardiomyopathy, and relate it to cobalt toxicity and beriberi. Recognize alcohol itself as a controversial cause of cardiomyopathy.
Mention the typical setting for peripartum cardiomyopathy, and explain why we suspect a nutritional deficiency.
Recall that disarray of the myocardial fiber arrangement as the typical, though not invariable, feature of hypertrophic cardiomyopathy. Recognize "asymmetric septal hypertrophy" and "idiopathic hypertrophic subaortic stenosis" as the classic hypertrophic cardiomyopathy in which the septum is primarily involved. Recognize "obstructive hypertrophic cardiomyopathy" as the feared consequence of an over-thick septum, and describe this syndrome. Cite the gene responsible for many of these cases.
Briefly describe the heart disease seen in sarcoidosis and systemic amyloidosis, and recall the prevalence of minor amyloid deposits in the hearts of the elderly.
Describe endomyocardial fibrosis as seen in the apices of hearts of young Africans. Describe Loeffler's endocarditis ("with eosinophils") clinically and histologically. Describe endocardial fibroelastosis as seen in U.S. infants, both grossly and clinically.
Describe cardiac damage from anthracyclines (adriamycin and its relatives), and from cocaine.
Recall ruptured MI, penetrating injury, and backwards rupture of an aortic dissection as the only common causes of hemopericardium.
Tell how much fluid is required to produce cardiac tamponade, and under what circumstances it must accumulate.
Recognize the causes of pericarditis from table 13-9 of "Big Robbins". Mention the classic posture assumed by patients with pericarditis. Mention some of the organisms (TB, viruses) that may come from a serous pericardial effusion.
Recognize myocardial infarcts, uremia, radiation, lupus, rheumatic fever and trauma as the causes of fibrinous pericarditis, describe the origin of the distinctive physical sign, give the gourmet comparisons, and mention the anatomic progression and clinical prognosis.
Describe the causes and outcome of purulent (suppurative) pericarditis. List the significant causes
of hemorrhagic pericarditis (i.e., TB![]() and cancer) and
caseous
and cancer) and
caseous ![]() pericarditis (TB).
pericarditis (TB).
Recognize the cancers that tend to metastasize to heart. Be aware of the problems that such metastases can cause, and the difficulty of making the diagnosis.
Recall atrial myxomas ("wrecking balls") as the only common primary tumors of the heart. Tell where they arise and how they cause problems. Recognize their gross and microscopic appearances.
Recognize any good example of each of the types of lesions depicted in the videodisc series.
Say "REE-nin", not "RENN-in", when talking about that important hormone from human physiology. Rennin is from a calf's stomach and you use it to make cheese.
The heart has its reasons of which Reason knows nothing. -- Blaise Pascal
The heart weeps for what is has lost; the spirit rejoices for what it has found. -- Sufi Proverb
![]() {03467} normal histology
{03467} normal histology
INTRODUCTION
Cardiac pathology is relatively straightforward, if you understand the heart's physiology.
There are only a few important diseases and patterns of injury, and most of these are fairly well understood.
Myocardial biopsy is becoming more and more important, especially in the transplant era. All about it: Mayo Clin. Proc. 86: 1095, 2011.
Looking at pictures of the heart? Remember:
* The histology textbooks usually do not mention the fat cells that one may see in the myocardium; this correlates with visceral obesity, as does excess fat around the heart (J. Clin. Endo. Metab. 98: 1189, 2013).
![]() I think that the pathology of the heart presents fewer difficulties than any other organ system except
GI tract, as long as you understand the physiology.
I think that the pathology of the heart presents fewer difficulties than any other organ system except
GI tract, as long as you understand the physiology.
* We used to think that myocardial cells were post-mitotic. During the 1990's, it became clear that hyperplasia is a feature of the deformity in the vicious cycle of congestive heart failure. Now it's clear (from studies of nuclear fallout) that we regenerate maybe 1% of our heart fibers yearly (Science 324: 98, 2009). Update Mayo Clin. Proc. 88: 871, 2013.
A variety of genetic syndromes produce various problems with the cardiovascular system. I have tried to resist the tremendous temptation to describe all my favorites. Instead, I've included only the ones that a generalist should know.
 * THE PROARRHYTHMIAS FIASCO
* THE PROARRHYTHMIAS FIASCO
Had CAST not included a placebo group, the erroneous conclusion that drug therapy did no harm might have been reached.
--NEJM, cited above.
"Proarrhythmias" are rhythm disturbances generated or made worse by anti-arrhythmic drugs. And they are quite common. In past decades, there have been fads for prescribing anti-arrhythmic drugs to asymptomatic people with ordinary ventricular ectopic beats (PVC's) and who have never had a heart attack. This is indefensible (Am. J. Card. 64: 50-J, 1989; NEJM 312: 193, 1985), and we can only guess how many thousands of people have died worldwide as a result.
|
|
|
ATHLETE'S HEART (is good: Eur. Heart. J. 17: 127, 1996; making the call Prog. Card. Dis. 54: 387, 2012).
![]() Athletic heart
Athletic heart
Tom Demark's Site
The heart is special, because one of the most common "abnormalities" is the desirable result of vigorous aerobic training.
|
The athlete's heart is hypertrophied, often remarkably so.
Even supine, you can often feel the apex beat far lateral to
the mid-clavicular line. The pulse is slow (50-60 beats at rest), and the QRS complex often
tremendously large (why?).
* An athlete's hypertrophied ventricles also produce much more atrial natriuretic factor than a couch potato's, helping dispose of a sodium load -- perhaps this is why exercise helps patients with hypertension. * The right ventricle, of course, is often big as well (Prog. Card. Dis. 54: 397, 2012). Further, the athlete develops tremendous collateral circulation. If something happens to one coronary artery, the percentage of lost myocardium (if any) will be less. There will also be more total heart muscle remaining. If there is serious ischemia of a portion of heart, surviving a major rhythm disturbance may still be a problem, but death from cardiogenic shock in the acute phase is far less likely. |
 Maurice Greene comes from my home town |
Also, exercise does offer some protection from coronary artery atherosclerosis. Most runners also avoid tobacco and cholesterol-raising foods, and exercise tends to keep hypertension and adult-onset diabetes at bay, making it harder to sort out the benefit of exercise itself. Nevertheless, even the best runners enjoy no absolute immunity to coronary artery atherosclerosis, with all its serious side-effects.
People who weren't thinking (including some physicians in the 1950's) used to talk about "athlete's heart syndrome" as if it were something to be avoided. The "reasoning" was that failing hearts in disease tend to be hypertrophied, and.... Silly, okay. Probably the only common situation in which a person should avoid heavy aerobic training is some birth defects in which cardiac hypertrophy is likely to impair outflow from the left ventricle. (Outstanding among these is hypertrophic cardiomyopathy).
* One group reports that athletes' hearts do not hypertrophy to a thickness of more than 12 mm unless their chambers are also dilated, which should help make the distinction from diseased hearts: J. Am. Coll. Card. 40: 1431, 2002; this surprises me, as it does not square with my own experience after having autopsied a few high-performance athletes.
Your lecturer agrees with the position taken in Lancet 371: 1489, 2008 that neither routine ultrasonography nor routine EKG's for all athletes are worthwhile -- they will generate much confusion and expense and save very few lives.
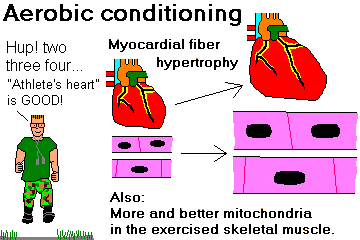
You'll need to know this somewhat artificial classification of cardiac hypertrophies, since a clinician or radiologist may ask you about it, and as there's talk about "remodelling of the heart" (NEJM 358: 1370, 2008), it may eventually come down to a real science.
ECCENTRIC HYPERTROPHY: The heart is big and it is NOT able to empty properly. The cardiac myocytes are LONGER, with new sarcomeres laid down BEYOND existing ones. Thick wall, chamber is very expanded and does not empty adequately. Perhaps the heart is pumping too much blood (anemia, AV shunts, thyroid disease, others) and/or it refills (aortic regurgitation) and/or there's a dead zone from an old healed infarct and/or it's doing its best but can't keep up for whatever reason (i.e., congestive heart failure from most causes). Looks bigger than a concentrically hypertrophied heart of the same weight on a chest x-ray. (Why?) Framingham confirms that concentric hypertrophy is linked to a good ejection fraction and eccentric hypertrophy to a reduced ejection fraction: Am. J. Card. 113: 117, 2014 -- no surprise at all.
PHYSIOLOGIC HYPERTROPHY: Aerobic athlete; also late in pregnancy. Thick wall, the chamber can fill tremendously but empties very well. Word on the street is that hypertrophied cells are longer than they are thicker, but since so few specimens come in for study, the question's not settled. Watch for the molecular biology -- including more expressions of variant myosins -- to be uncovered as we learn more about conditioning.
HYPERTROPHIC CARDIOMYOPATHY: Uneven fiber enlargement and scrambling not to be confused with any of the above. Bumps on the heart muscle notably around the aortic outflow track.
JAMA reports on deaths during triathlons (risk 1.5 per 100,000 per tri) confirms for me that "enlarged heart" in fit people without hypertrophic cardiomyopathy kills no one, though the authors reached a different conclusion. Of 14 death, 13 look like drownings, and one triathlete fell off the bicycle and broke his neck. Despite the drowning victims having "cardiovascular abnormalities" ("six had mild left ventricular hypertrophy".... they are triathletes, go figure), nobody dropped dead from anything resembling heart trouble during the running or biking. See JAMA 303: 1255, 2010. This fascinating article also confirmed for me that no one knows the "normal weight" for the heart of an elite athlete / hard physical laborer, since so few die. For me, the question of when a medical examiner can say "the death was due to an enlarged heart" remains unanswered.
Measurements for future pathologists:
350 gm... Traditional normal upper limit of weight for a (slim adult couch potato's) heart
1.5 cm... Traditional normal upper limit of thickness for a (slim adult couch potato's) left ventricle
0.5 cm... Traditional normal upper limit of thickness for a (slim adult couch potato's) right ventricle
Measurements don't include the trabeculae carnae.
Fun to know: If the heart was once very hypertrophic but is so no longer (i.e., an athlete gone to seed, a hypertensive or valve-disease patient successfully treated), the anterior and posterior descending coronaries are very wiggly "accordion arteries". Why? I've seen this often.
When someone dies suddenly with only a large heart -- concentric or eccentric hypertrophy, and the medical examiner finds no other cause of death, it's considered acceptable to blame a rhythm disturbance (NEJM 358: 1370, 2008 -- the article makes the point that in athletic/physiological hypertrophy, you do NOT get rhythm problems as a result). I've always been shy about doing this, but it comes up often in high-profile "sudden deaths of athletes." In my 1300+ autopsies, I have never had to invoke "enlarged heart" as cause of death.
CONGESTIVE HEART FAILURE ("CHF"; update Lancet 373: 941, 2009)
Inability of the heart to handle the volume of blood returned to it.
Either the heart muscle cannot pump because of intrinsic disease, or the blood is flowing in the wrong way, or the heart must pump against excessive resistance, or the heart must pump a preposterously large amount of blood (the latter is "high output failure").
Physiologists speak of "forward failure" (i.e., inability to perfuse the arteries, notably the kidneys to dispose of a sodium load) and "backward failure" (i.e., congestion and its problems). Both occur simultaneously, of course, but one or the other may be more obvious clinically.
The distinction between "congestive heart failure" and "cardiogenic shock" is admittedly artificial. "Cardiogenic shock" is a term reserved for the acute situation (usually a myocardial infarct); "failure" can simply mean inability to handle the ordinary venous return.
 As the heart is forced to work extra-hard, it undergoes HYPERTROPHY (i.e., more muscle
mass) and perhaps dilatation (i.e., chamber enlargement, which helps pump the blood; remember Starling's
Law?) Eventually, however, the heart's strength cannot increase further, and the organ appears to
give up (i.e., it stops "obeying Starling's Law"). Now the chamber does not
empty fully.
As the heart is forced to work extra-hard, it undergoes HYPERTROPHY (i.e., more muscle
mass) and perhaps dilatation (i.e., chamber enlargement, which helps pump the blood; remember Starling's
Law?) Eventually, however, the heart's strength cannot increase further, and the organ appears to
give up (i.e., it stops "obeying Starling's Law"). Now the chamber does not
empty fully.
Exactly why the over-burdened heart's strength starts to fail is often unclear, and its response to pharmacologic interventions often makes the picture more mysterious. There is talk of induction of an abnormal myosin isoenzyme which is a poor ATP-ase, decreased numbers of beta sympathetic receptors, etc.
There's much interest in the effects of heart failure itself on the heart, both changes in the shape of the heart that render its pumping and/or filling less effective ("remodelling"; Am. Heart J. 130: 153, 1995), and problems with the cells themselves (notably failure of reuptake of calcium from the sarcoplasmic reticulum; see Am. Heart J. 129: 684, 1995). The "Batista partial left ventriculectomy", in which some muscle is actually removed from the failing heart, is now common, though results are mixed. (Randas Batista was a heart surgeon in a tiny town in rural Brazil; his success in 1996 probably wasn't "ethical" by our standards, but the procedure's in use anyway.) The "dynamic cardiomyoplasty", in which muscle from the back is wrapped around the heart and given a pacer, and the metal mesh "heart sock" are both designed to help keep the ventricle shaped normally.
* I wasn't at all surprised to learn that in an 2008 autopsy series (Arch. Path. Lab. Med. 132: 1392, 2008), every one of 70 obese decedents had heart weight "above normal." Why is the heart of a fat person bigger? Is it simply from the extra exercise of carrying around 100-200 or more pounds of weight? ( think so -- "the simplext explanation is usually the best".) Or is it the result of lack of adiponectin secretion by overstuffed adipocytes (Nat. Med. 10: 1384, 2005)? How can anybody tell? Anyway, after bariatric surgery, at least in the young, the heart tends to return to normal size (J. Am. Coll. Card. 51: 1342, 2008).
You can help out a congestive-heart-failure person, somewhat, by improving aerobic muscle tone (i.e., more efficient burning of fuel), but exercise is no panacea (J. Am. Coll. Card. 25: 1239, 1995; J. Am. Coll. Card. 27: 140, 1996; JAMA 283: 3095, 2000).
* Future pathologists: Serum cardiac troponin T (late 20th century pre-hospital to screen for MI's: Am. Heart J. 138: 45, 1999) as a marker for how bad my congestive heart failure is today: Am. Heart J 138: 95, 1999.
Nesiritide, a natriuretic peptide originally found in brain, helps CHF: NEJM 343: 246, 2000; you can also measure levels to detect CHF (NEJM 347: 161, 2002); B-type natriuretic peptide triumphs as a way to distinguish CHF from other causes of dyspnea: NEJM 350: 647, 2004. No surprise.
* Watch galectin-3, both as a molecule produced by macrophages in the failing heart that causes the fibroblasts to make collagen, as a serum marker for severity of congestive heart failure, and as a target for therapy.
|
|
LEFT-SIDED CONGESTIVE HEART FAILURE
Failure of the left side of the heart to pump sufficient blood.
Except in the case of pure mitral stenosis (why?) or amyloidosis (why?), the left ventricle will be hypertrophied and dilated. The left atrium will usually be, also (and especially in mitral valve disease, why?)
THE COMMON CAUSES OF LEFT-SIDED FAILURE
Ischemia (old or recent myocardial infarct, ischemic muscle disease)
Aortic or mitral valve disease
Systemic hypertension
Myocardial disease / cardiomyopathy
NOTE: Of these, uncontrolled "systemic hypertension" (i.e., too much blood to push through too-narrow arterioles) is the most common; when the heart fails, blood pressure drops, making the true cause less obvious. See JAMA 273: 1363, 1996 (Framingham). How it progresses: JAMA 275: 1557, 1996; J. Am. Coll. Card. 25: 888, 1995.
THE COMMON EFFECTS OF LEFT-SIDED FAILURE
First, on exertion
Later, PAROXYSMAL NOCTURNAL DYSPNEA ("cardiac dyspnea"); on lying down for a while, fluid redistributes itself in the body, resulting in pulmonary edema. I think that the reason that it's paroxysmal (i.e., comes on all of a sudden) is that as the lungs become heavier (i.e., congestion, maybe edema) their weight presses on the pulmonary veins which in turn makes them more congested. Patients may throw the windows open at night, or learn to sleep on various numbers of pillows; you the physician will hear rales; the pathologist may see "brown induration" and hemosiderin-laden "heart failure" macrophages; remember these?
DIASTOLIC HEART FAILURE is a special situation in which the ejection fraction is normal but the person is still in failure. The ventricle will not relax / is too stiff to fill properly. Hence, diastolic volume is low. It is not rare; the pathophysiology is being worked out (NEJM 350: 1953, 2004). Death may result from a sudden gush of pulmonary edema fluid. Don't overdiagnose it... remember that in dilated cardiomyopathy, the overstretched wall of the heart may be stiff.
HIGH-OUTPUT FAILURE is a special situation in which the heart fails because the body demands more blood than it can pump. You'll see dependent edema probably because the veins of the body constrict extra-hard to return blood to the heart. The causes:
Anemia
Hyperthyroidism (little arteries dilate throughout the body)
High fever
Shunts between an artery and a vein
Beriberi (poor autonomic control)
Paget's disease of bone (abnormal bone vasculature)
Iatrogenic (i.e., shunts in dialysis)
RIGHT-SIDED CONGESTIVE HEART FAILURE
Failure of the right side of the heart to pump enough blood.
As you'd expect, the right ventricle and atrium will usually be hypertrophied and dilated.
THE COMMON CAUSES OF RIGHT-SIDED FAILURE
Pulmonary emboli (acute or chronic)
Any disease interfering seriously with lung ventilation
Emphysema
Cystic fibrosis
Fibrosing lung
Most others
NOTE: The mechanism, of course, is increased pulmonary vascular resistance (due to fibrosis and/or the hypoxic vascular response; remember this?)
Left-sided heart failure!
Cardiac defects with left-to-right shunts (why?)
THE EFFECTS OF RIGHT-SIDED FAILURE
Splanchnic congestion (you'll feel big livers & spleens; check for "hepatojugular reflux")
Jugular venous distention (look carefully)
Edema in the feet and elsewhere (from increased venous hydrostatic pressure, etc.)
Effusions (transudates, of course; notably pleural, notably more on the right side than on the left; why?)
NOTE: "Cardiac cirrhosis" of the liver, often discussed in textbooks as the result of right-sided failure, almost never happens. The one time you might see it is in longstanding, severe tricuspid insufficiency, with or without right-sided failure (why?) If you are this sick, "cardiac cirrhosis" is probably the least of your worries.
NOTE: Some pathophysiologists include cardiac tamponade as a type of right-sided failure.
* Good news: In contrast to studies of selected patient populations with various illnesses from decades ago, black and white people with congestive heart failure seem to get equally good treatment (JAMA 289: 2517, 2003). This seems to be part of a general trend to eliminate (and even reverse) the past tendency to undertreat minority patients (NEJM 354: 1147, 2006).
NOTE: Especially with new biotech products, watch for more aggressive treatment of anemia of chronic disease as a way of helping your CHF patients (Am. Heart. J. 155: 751, 2008).
ISCHEMIC HEART DISEASE
|
|
The cause of around 750,000 deaths annually in the U.S. In at least 90% of the cases, the problem is coronary artery atherosclerosis (ASCVD).
In the setting of acute ischemia, one common mechanism of death is CARDIAC RHYTHM DISTURBANCES ("arrhythmias", one of the great misnomers in medicine). Don't worry about the details here; just remember what you've already learned about (1) ischemia making membranes abnormally permeable to ions, and (2) action potentials and how they result from altered permeability to ions.
Cigaret![]() smoking is a risk factor for coronary atherosclerosis, and also
sensitizes the myocardium to be susceptible to rhythm disturbances in the setting of ischemia.
smoking is a risk factor for coronary atherosclerosis, and also
sensitizes the myocardium to be susceptible to rhythm disturbances in the setting of ischemia.
It's also worth remembering that coronary arteries usually increase their diameters substantially as atherosclerosis worsens (study from my old department at Bowman-Gray: JAMA 271: 289, 1994), a phenomenon that saves lots of lives.
Future pathologists: We (unlike angiographers) refer to coronary artery stenosis in terms of percentage of cross-sectional area occluded. A pathologist's 75% occlusion is an angiographers 50% occlusion. A pathologist's 90% occlusion is an angiographer's 70% occlusion. Why?
You know the dominant coronary artery is whichever supplies the posterior descending coronary artery.
ANGINA PECTORIS: Pain in the chest from coronary insufficiency, in the absence of myocardial infarction
Regardless of its category, all angina is due to some combination of coronary stenosis (usually atherosclerotic), coronary spasm (demonstrable on angiogram), thromboxane A2 release and platelet aggregation, and temporarily increased myocardial work load.
STABLE ("classic", "typical", "Heberden") ANGINA generally results from increased work in a patient with coronary atherosclerosis, and relieved by rest.
Generally, three-vessel disease with >=75% cross-sectional stenosis in each of three coronary arteries is sufficient to cause problems. Of course, finding 90+% stenosis is commonplace in the U.S. Ask patients about exacerbation of pain on climbing stairs or walking against cold wind.
UNSTABLE ("pre-infarction", "crescendo", "accelerated", "acute coronary insufficiency") ANGINA
In most cases, this is probably due to a thrombus developing, by fits and starts (white regions, organization, etc.), over a ruptured plaque. Untreated, many of these people get an MI soon.
* "Is it really a heart attack?" On rotations, you'll learn that folks with ST-segment elevations probably have thrombi and get sent for thrombolytic therapy and/or a stent. Those with nonspecific T-wave changes or ST-segment depression, or with acute chest pain suggesting myocardial ischemia but no EKG changes will be classified as having "unstable angina" (troponin not up) or "non-ST-segment-elevated myocardial infarct", where fibrinolysis may not be the best treatment. The terminology may change again. It's fairly common, especially in women, to have EKG-and-enzyme myocardial infarction without angiographically-demonstrable vessel obstruction (i.e., no vessel is narrowed anywhere more than 50%); we used to say "Prinzmetal's" but now it's clear using ultrasounds and isotope scans at least half have an ulcerated plaque that we may suppose is atheroembolizing (Circulation 124: 1414, 2011). Sometimes it's tough to know, especially without an autopsy. "Infarctlets" / "CK leaks" / "troponin-positive acute coronary syndrome" are now discussed as being coronary ischemic events that raise cardiac enzymes but do not produce the EKG changes of a "true MI". No one knows exactly what to do with these patients (Br. Med. J. 324: 377, 2002; the traditional rx of calcium channel blockers fails more often than not Chest 123: 380, 2003). Now it does seem that implementing a sensitive troponin I assay and treating folks who are positive as heart attack patients does prevent deaths during the following year (JAMA 305: 1210, 2011).
* The endothelial nitric oxide synthetase gene has a mutant allele that is a strong predictor for coronary artery spasm: Circulation 99: 2864, 1999. Updates -- these people are easily treated and extremely satisfied Am. J. Card. 105: 792, 2010; Trans. Res. 162: 64, 2013.
* A "mouse model" (?) for Prinzmetal's has a mutation in a minor potassium pump; sudden cardiac death and coronaries that over-react to minor vasoconstrictors characterize this mouse: Nat. Med. 8: 466, 2002; J. Clin. Invest. 110: 203, 2002.
CARDIAC SYNDROME X ("microvascular angina"), with classical clinical angina and wide-open coronary arteries, and a generally good prognosis (seldom or never causes death), is an autonomic (?) disturbance in which the smooth muscle of blood vessels does not dilate appropriately and/or constricts too easily.
These people may not get the red flush on re-perfusing a forearm made ischemic by a blood pressure cuff.
More about this arcane syndrome, which is quite common and seems to have something to do with insulin resistance, in Lancet 342: 136, 1992, and Am. J. Card. 79: 961, 1997; Lancet 351: 1165, 1998; NEJM 346: 1948, 2002; Hosp. Pract. 35(2): 75, Feb 15, 2000.
We're only starting to make sense of the histopathology, and so far it's either common-sense anatomy or "normal vessels that must be reacting abnormally." "Coronary microvascular disease" and how difficult it is to study: NEJM 356: 830, 2007 -- causes include the familiar arteriolar sclerosis of hypertension, the strange muscling of the little arteries in hypertrophic cardiomyopathy, a poorly-understood change caused by aortic stenosis, and Fabry's (extreme; Heart 92: 357, 2006).
Don't confuse it with METABOLIC SYNDROME X, which is the poorly-understood, all-too-common syndrome of obesity, hypertriglyceridemia, low HDL, hypertension, and insulin resistance. Of course, the two may coexist.

|
|
|
|
|
|
|
|
|
|
|
This common (maybe 1 million/year in the U.S.) catastrophe underlies many, but by no means all, fatal cases of ischemic heart disease.
There are fewer myocardial infarcts nowadays than in the past, and the odds for a patient with a myocardial infarct are much better, too, especially once he or she has made it to the hospital (NEJM 334: 884, 1996). Update: The numbers are still falling dramatically, with one emergency room having 133 cases of ST-segment elevation MI in 1999 and only 50 cases of ST-segment elevation MI in 2008 (NEJM 362: 2155, 2010).
CAUSES OF MYOCARDIAL INFARCTS
ATHEROSCLEROSIS: Makes up 90+% of coronary artery disease
{03476} atherosclerosis, coronary artery
{06531} ruptured plaque with thrombus
|
|
|
|
The pathologist can usually find either a RUPTURED PLAQUE (often with an overlying thrombus, hence the archaic name "coronary thrombosis"; review of coronary thrombi Am. J. Card. 68: 28B, 1991), or (less often) a HEMORRHAGE INTO A PLAQUE, ballooning its cap against the opposite wall. If neither are present, but there's horrendous atherosclerosis and no other explanation, we assume the thrombus lysed.
No surprise: The Armed Forces Institute of Pathology documents that sudden cardiac death occurring during intense physical exertion typically results from a ruptured plaque: JAMA 281: 921, 1999.
The "culprit lesion" identified by the pathologist at autopsy was often a small one previously; the danger posed by an atheroma has just as much to do with its ability to rupture and/or experience hemorrhage into itself (think "fatal heart attack") as it does to cause stable stenosis (think "stable angina"). Clinicians are now coming to understand this (NEJM 364: 226, 2011).
Long afterwards, look for a recanalized thrombus.
*Space-age medicine! Viewing the thrombus by angioscopy NEJM 326: 287, 1993. Unstable angina thrombi are likely to be white fibrin-platelet thrombi or organizing thrombi (why?), etc. And the thrombi overlie plaques that are lipid-rich and/or disrupted (no surprise): Am. J. Card. 79: 1106, 1997.
The recreational drug (1) produces coronary artery constriction (spasm, or whatever, nobody really
understands it NEJM 333: 1267, 1995; Am. J. Card. 79: 492, 1997) and cardiac ischemia
and even infarction (Circulation 99: 2737, 1999),
especially when combined with cigaret![]() smoking (NEJM 330: 454, 1994), which is bad because both
increase the heart's need for oxygen; (2) makes the heart more prone to rhythm disturbances, perhaps
by enhancing the effects of endogenous catecholamines; (3) can produce single-fiber necrosis/apoptosis
and
contraction bands (something to do with ion channels), perhaps leading to myocarditis and/or dilated
cardiomyopathy. It is also known to produce (4) a dilated
cardiomyopaty (Lancet 369: 1574, 2007).
(5) The fibrous-hyaline thickening of the walls of the small arteries of the hearts
of cocaine users is unexplained but seems to be a real effect (Eur. Heart J., Jan 12 2010).
smoking (NEJM 330: 454, 1994), which is bad because both
increase the heart's need for oxygen; (2) makes the heart more prone to rhythm disturbances, perhaps
by enhancing the effects of endogenous catecholamines; (3) can produce single-fiber necrosis/apoptosis
and
contraction bands (something to do with ion channels), perhaps leading to myocarditis and/or dilated
cardiomyopathy. It is also known to produce (4) a dilated
cardiomyopaty (Lancet 369: 1574, 2007).
(5) The fibrous-hyaline thickening of the walls of the small arteries of the hearts
of cocaine users is unexplained but seems to be a real effect (Eur. Heart J., Jan 12 2010).
Future pharmacologists: The drug opens sodium channels, perhaps opens calcium channels, and prevents synaptic re-uptake of catecholamines. Crystal meth probably does the same thing (J. Tox. 41: 981, 2003).
* Hopefully no one was surprised by the results of a huge study that showed that cocaine's effects on the heart are not mediated by its causing precocious atherosclerosis (Am. Heart. J. 150: 921, 2005).
PRINZMETAL'S CORONARY SPASM
MYOCARDIAL BRIDGE and DIVING CORONARY ARTERY, especially involving a portion of the left anterior coronary artery.
VASCULITIS
Remember (1) lupus; (2) polyarteritis nodosa; (3) rheumatoid arthritis; (4) Kawasaki's; (5) Takayasu's; (6) mycotic aneurysms (remember what those are? seen in bacterial endocarditis); (7) rarely, exotic infections.
* Coronary artery aneurysms suggest Kawasaki's or Behcet's; they are known in polycystic kidney disease.
{06569} polyarteritis nodosa of a coronary artery
{06587} aspergillus![]() infection of a coronary
artery
infection of a coronary
artery
|
|
EMBOLIZATION (i.e., bacterial endocarditis)
SYPHILIS![]() (classic!)
(classic!)
{03525} syphilis. Nice plasma cells.
DISSECTING HEMATOMA
A "dissecting aneurysm" can slide right up through a coronary ostium and cause occlusion. Or (and this is fairly well-known, especially shortly after childbirth) a coronary artery can spontaneously develop a dissecting hematoma (Am. J. For. Med. Path. 33: 26, 2012), or blunt trauma to the chest can cause a coronary artery dissection ("traumatic myocardial infarction": Heart & Lung 41: 294, 2012).
{06575} coronary artery dissection
SHOCK and LEFT-SIDED FAILURE, even if mild, in the setting of serious coronary artery stenosis.
A drop in blood pressure from any cause is likely to produce a SUBENDOCARDIAL INFARCT. (By definition, a subendocardial infarct is less than 50% as thick as the wall). This is a watershed infarct of the myocardium farthest from the coronary, but not close enough to the chamber to get its nutrients from the blood in the chamber.) If all three coronaries are stenotic, the infarct is likely to be circumferential.
* A favorite place to find subendocardial infarcts is in the tips of the mitral valve's papillary muscles. Why?
* Truth is, in a myocardial infarct, the injury and the death begin in the subendocardium whether or not it progresses to transmural involvement.
* Amyloid of the coronaries shouldn't cause an infarct, as the lumen is open and the endothelium undamaged.
{03386} coronary artery amyloidosis
{06584} amyloid (special stain)
{17483} amyloid myocardium
CILNICAL PICTURE OF THE "MI" PATIENT
Everyone knows the "typical" uncomplicated heart attack victim. There is chest pain (maybe, perhaps "crushing") radiating to the (left arm? jaw? abdomen? wherever?), perhaps with diaphoresis, perhaps with shortness of breath, perhaps with a feeling of fear, or perhaps with none of these ("silent infarct").
Serum enzymes (troponin, CK, perhaps with an "MB" cardiac isoenzyme band; LDH is of historic interest only) begin to rise in perhaps 2-4 hours, but may be normal in a day or two. Ask your cardiologist how often to check. Lowering the threshold for "a positive troponin" in the setting of likely MI seems to save lives: JAMA 305: 120, 2011.
The EKG changes depend on the location. Remember the subendocardial infarcts are notoriously hard to pinpoint by EKG, and that posterior wall infarcts are easy to miss, too.
You'll learn of the management and treatment of these patients while in the "unit". Remember that education about reducing risk factors is an important part of cardiac rehabilitation.
* Speaking of "cardiac rehabilitation": As a med student, I watched heart-attack survivors participate at great expense in kindergarten-level exercise programs in special hospital areas, supervised by boarded cardiologists. Not surprisingly, this got replaced (under the much-maligned "managed care") by expenditures on prevention (Am. J. Card. 79: 1075, 1997); for the "medically indigent" who need to change their lifestyles, good results are obtained simply by talking and explaining nicely (Am. J. Card. 79: 281, 1997).
* Splitters: You may learn this subclassification of MI's, which guides treatment....
PATHOLOGY
0-30 minutes
Wavy fibers at the edges, loss of glycogen from cytoplasm.
1- 2 hours
Mitochondrial calcium, maybe contraction bands, maybe hydropic changes, maybe even a little fatty change.
4-8 hours
Earliest nuclear changes, polys appear; you may see a bit of dark mottling grossly; any time there's been reperfusion you can see hemorrhage
8-24 hours
First clear gross changes, i.e., pallor; good
coagulation necrosis![]() ;
often good contraction bands;
definitely feels soft by 24 hours
;
often good contraction bands;
definitely feels soft by 24 hours
24-72 hours
Looks very inflamed and injured under the microscope, lots of polys, fibers are obviously necrotic; infarct feels soft and looks pale and yellowish (why?)
3- 7 days
Macrophages, granulation tissue starts at rim; grossly you see the red granulation tissue around the infarct
10 days
Nice granulation tissue; macrophage cleanup team may be removing the dead fibers
2-6 weeks
Macrophages continue the cleanup as scar forms; necrotic muscle may persist but is not inflamed
7-8 weeks / 2 months
Nice scar. Healing is prettty much finished.
* Today's standard for autopsy reports: "Acute" means polys, "Healing" means the polys are gone but there are macrophages, "Healed" means the macrophages are gone. "Microscopic" means less than 10% of the left ventricle (sic.), medium is 10% to 30%, large is more than 30% (Am. Heart. J. 144: 957, 2001).
* Future radiologists: We are developing scans that light up just the macrophages, to see how big the healing infarct is (Radiology 264: 428, 2012).
|
|
|
|
|
|
Among these, the only items that may be unfamiliar are WAVY FIBERS (they had stopped beating and were roughed-up by the beating of the rest of the heart) and CONTRACTION BANDS ("myofibrillar degeneration"; densely eosinophilic cross-bands that probably result from calcium entering membrane-damaged cells during reperfusion, i.e., REPERFUSION INJURY. Remember that? -- We still are searching for effective treatments that will keep injured myocardial cells safe while they are being reperfused J. Clin. Inv. 123: 92, 2013)
* The AFIP has finally documented what real-world pathologists have known and used for decades: contraction bands let you know that a sudden death is of cardiac ischemic origin (Lancet 347: 1710, 1996).
* Future pathologists also note: Contraction bands can probably result from epinephrine administration and/or electric shocks in CPR. We know that they are common enough in deaths from lightning strikes.
NOTE: Classically, the coronary arteries have the following distribution, and their occlusion will result in TRANSMURAL (across-the-wall, or at least more than 50%) infarcts in the corresponding distribution
Right: Posterior-inferior wall, posterior 1/3 of septum
Left anterior descending: Anterior wall, anterior 2/3 of septum
Left circumflex: Lateral wall
There's plenty of variability. Especially when there's atherosclerosis, collateral formation may result in the "best" artery supplying most of the heart, with minor occlusions producing "infarction at a distance". Don't worry yet about "which gives you a bundle branch block", etc., etc.
NOTE: Infarcts almost never involve the right ventricle, unless it is extremely hypertrophied (why do you think?) If the infarct EXTENDS as a result of more mayhem in the coronaries, expect a mix of ages.
* Future pathologists: Try the NITROBLUE TETRAZOLIUM technique to demonstrate early myocardial infarcts. Drop a slice of heart in the solution, and viable heart, containing an oxidizing enzyme, will stain brown, and dead heart remain pale. I could never get this to work.
* Future pathologists: Don't mistake livor mortis for a posterior wall MI!
* Tomorrow's pathologists: The postmortem MRI scan can help with the diagnosis of myocardial infarction. Myocardial infarcts more than one hour old show well on both MRI and on prosection. MRI may actually be superior to the gross exam for MI's with death during the first hour (J. Am. Coll. Card. 62: 617, 2013.) One limit on this study is that both imaging and pathologists paid little attention to the coronary arteries themselves.
{10103} myocardial infarct, acute
{06639} very early MI (bottom only)
{06428} myocyte degeneration (hydropic change)
{06431} myocyte degeneration (hydropic change)
{06642} contraction bands
{06651} contraction bands
{06645} necrosis and polys
{06630} subendocardial MI
{06654} good necrosis and polys (NOTE: the myocyte nuclei are
homogenized rather than pyknotic; that still means "dead")
{06443} road-kill, lots of polys, fibers very dead; good contraction bands remain
{06446} nice granulation tissue
{06663} nice granulation tissue
{06666} nice granulation tissue (left)
{06449} nice scar
{06338} nice scar
{06455} nice scar (trichrome)
COMPLICATIONS occur in many but not all myocardial infarcts.
RHYTHM DISTURBANCES may begin at any time until the damage to the conduction system is healed. Formerly the great killer of "MI" patients, pharmacologic therapy is now generally successful in managing these.
LEFT-SIDED CONGESTIVE HEART FAILURE results from extensive damage to the heart. Whether or not there's been a known episode of infarction, a person with severe coronary disease can get intractable heart failure on the basis of ischemic scarring.
CARDIOGENIC SHOCK results from necrosis of more than >=40% of a non-athlete's myocardium. This is usually fatal.
RUPTURE (ouch!) of the heart may occur, typically when the damaged heart is most soft (days 3-7 or thereabouts), but day 1 ruptures are not unheard-of.
Rupture of the FREE WALL will result in hemopericardium, tamponade, and instant death.
Rupture of the SEPTUM will result in a sudden left-to-right shunt.
Rupture of the PAPILLARY MUSCLE produces severe mitral regurgitation.
{03614} ruptured wall
{07141} hemopericardium
{03617} ruptured septum
{53285} ruptured septum
ANEURYSM FORMATION, MURAL THROMBUS FORMATION, AND EMBOLIZATION are dread, common side-effects of myocardial infarcts. Ventricular aneurysms begin with the paradoxical movement of the necrotic myocardium outward during systole; later the fibrous scar balloons. Large infarcts can produce large aneurysms that continue to balloon out. Having a big aneurysm following a myocardial infarct greatly interferes with pump effectiveness. Embolization from a mural thrombus (with or without an aneurysm) is often devastating.
{06323} myocardial ventricular aneurysm
![]() Ventricular aneurysm with thrombus
Ventricular aneurysm with thrombus
WebPath photo
* DRESSLER'S PERICARDITIS ("postpericardiotomy syndrome") is pericarditis (sometimes with life-threatening effusion) that supposedly occurs weeks to years after an MI or cardiac surgery. The sedimentation rate is elevated and there are variable reports of autoantibodies.
Dressler's is rare at best (Heart 80: 98, 1998), and some cardiologists don't believe in it (Angiology 47: 83, 1996).
Distinguish this late-onset illness (if it exists) from the pericarditis that's common during the first week following myocardial infarction just from the necrosis.
CHRONIC ISCHEMIC HEART DISEASE is cardiac muscle insufficiency due to scarring from old healed infarcts, not necessarily large or known to have occurred. It is a major cause of congestive heart failure. Think of this especially if your patient has nighttime symptoms but the heart is perhaps not enlarged.
SUDDEN CARDIAC DEATH
Definition: Death from cardiac causes in previously-asymptomatic person, within 1 (or 24) hours after onset of symptoms. Most often, the person feels odd, then falls over dead. Either there's a rhythm disturbance (most often, and typically "ventricular fibrillation"), or there's some sudden, severe outflow obstruction. (Of course, the "forme fruste" of sudden cardiac death is a "fainting spell"!)
You'll remember that "v-fib" results from widespread re-entry by whatever mechanism. If a ventricle is dilated (this usually happens in the right one), just having a long pathway does it. Decreased rate of conduction can result from ischemia, hyperkalemi, meds; short refractory period from sympathetic overload / epinephrine.
Every day, around 1000 people in the U.S. get "sudden cardiac death". (Some of these are probably included in the "1 million MI's" statistic; some probably aren't.)
Here's a rule: "Sudden death" means "sudden cardiac death". (Of course that includes pulmonary emboli.) Apart from extreme trauma, severing of a major body vessel, seizure death, electrocution, anaphylactic shock, super-fast poisons, or a hemorrhage that destroys the brain's medulla, there's probably nothing that can kill a human being in less than an hour that isn't on this list.
Here's another rule: There's almost always at least some warning in the weeks beforehand. See Circulation 114: 1146, 2006.
THE CAUSES OF SUDDEN CARDIAC DEATH
NOTE: Usually 75% concentric stenosis of all 3 coronaries, often more severe stenosis
If involvement of only one vessel is sufficient, at the textbooks tell you, it happens only in a small minority of these cases. The best study I've seen agrees (Hum. Path. 18: 485, 1987; still good.) Given the problems (not hektoening, which artery's dominant, etc., etc.) I remain unpersuaded that someone with 75% occlusion only at the widowmaker point can drop dead as a result unless something really exciting is going on in their life.
My former teammates Amy Kragel and her colleagues looked hard at the histopathology, and found that about 80% of these folks had a recanalized thrombus -- a surprising but credible finding (Am. Heart J. 127: 1588, 1994.)
Being stressed (epinephrine -- no wonder, see Lancet 370: 1089, 2007) and having tobacco on board probably exacerbate the rhythm disturbance, in ischemia due to atherosclerosis or anything else.
* Future whole-person-oriented medical examiners: When somebody drops dead with no pathology except three narrow coronary arteries, ask "Why today rather than yesterday?" You will almost always find out if you ask about the circumstances. For example, I hope no one was surprised that firemen are much more likely to have heart attacks while fighting fires than at any other time (NEJM 356: 1207, 2007 -- a firefighter seems to be almost equally in danger from the fire and from his coronaries).
Vasospasm
Fiber necrosis
Rhythm disturbances
For example, having only one (Pete Maravich had no left main, also J. For. Sci. 35: 981, 1990), or having one come off the pulmonary artery. These birth defects can cause other problems, too.
Ask a forensic pathologist about sudden death due to a "diving coronary artery", i.e., one which enters the myocardium too soon, and "myocardial bridging" (very common in hypertrophic cardiomyopathy and common enough in "normal people"), in which a band of heart muscle overlies a coronary artery (NEJM 339: 1201, 1998; Chest 116: 574, 1999).
Atrial myxoma or thrombus obstructing the mitral valve
Hypertrophic cardiomyopathy
Aortic valve stenosis from most any cause
NOTE: This is important. The mechanism is acute coronary insufficiency.
(1) The intra-myocardial branches of the coronary arteries fill only during diastole. In aortic valve stenosis, diastole is greatly shortened (why?).
(2) Bernoulli's principle (remember that?) results in blood being sucked out of the coronary arteries by the super-fast jet of blood passing through the narrowed aortic valve.
Cor pulmonale
Pulmonary embolus ("Do you think that should count as sudden cardiac death?"; maybe 50,000 extra "sudden death" cases among previously-healthy people in the U.S. per year)
Wolff-Parkinson-White, others ("bypass fibers", bundle of Kent; you'll learn about these "pre-excitation syndromes" on rotations; surgery Sci. Am. 269(1): 68, July 1993; gene NEJM 344: 1823, 2001)
Amyloid in the bundle of His (real frequency as cause of death is unknown, may be high)
Calcium / fibrosis / whatever as an acquired problem in the bundle of His (Lev's disease -- again, its real frequency is unknown)
Anti-Ro/SSA disease of the unborn and babies ("neonatal lupus").
* Other congenital heart block syndromes -- in children, expect to find calcification.
* NOTE: Since the conduction system is dissected and examined microscopically only when there is no obvious cause of death, the frequency of various "abnormalities" and whether they are related to death remains unknown ("narrowed / absent AV node artery"; "persistent fetal distribution", "ectopic AV tracts", more)
After surgically-induced trauma
Commotio cordis (myocardial concussion): Piezoelectric effect after a blow to the chest on top of the T-wave. See NEJM 362: 917, 2010. The current generation of chest protectors are inadequate. Special hazard for basketball, baseball catchers, hockey goalies, other activities: JAMA 287: 1142, 2002.
Recreational inhalant use (i.e., glue sniffing, etc.) sensitizes the heart to rhythm disturbances. This one's easy to miss, especially if the family or friends have removed the evidence. Finding this at autopsy is hard (Am. J. For. Med. Path. 27: 188, 2006); in fact, it's a famous cause of "negative autopsy" that you'll never solve.
Iron overload (rhythm disturbances)
| Myocarditis (even a little patch can cause rhythm problems and even death: "Hank Gathers's disease"); I "buy this" when you see necrotic myocytes along with a (T-)lymphocytic infiltrate ("Dallas criteria") |  Hank Gathers |
* Endocardial fibroelastosis (Am. J. For. Med. Path. 20: 357, 1999).
Ventricular septal defect involving bundle of His
* Familial syndromes with apoptosis of the sinus node and AV node (Circulation 93: 1424, 1996).
Right ventricular dysplasia (see below)
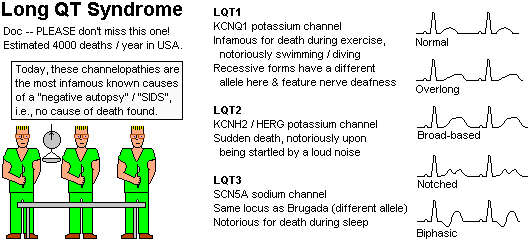
Channelopathies
The unusual EKG is not always expressed, so keep a high index of suspicion. Whenever you suspect long-QT, either on EKG or family history (i.e., unexplained sudden death, including SIDS, or an episode of torsade de pointes) or personal history (torsade, syncope), get consultation; genetic screening for suspected families will be routine probably by 2005. This business is very tricky. You'll learn on rotations what medications are contra-indicated in which syndromes, when to place a defibrillator, and so forth.
You shouldn't need to be reminded to do an EKG on all pre-sports physicals where there's a history of syncopal spells or sudden unexplained death in a family member under age 30.
Swimming seems to trigger sudden death when the mutation is in the potassium channel KCNQ1 (Mayo Clin. Proc. 74: 1088, 1999), while loud noises trigger when the mutation is in the potassium channel KCNH2 / HERG. When the mutation is in the SCN5A channel (same as a Brugada locus but a different allele), death is likely to occur during sleep.
Mayo's now finds that a third of unexplained sudden deaths are due to known channelopathies. Regrettably, Mayo's reports that paraffin-embedded material, which is all that's usually left long after an autopsy, doesn't seem to preserve these genes well. See Am. J. Clin. Path. 129: 391, 2008.
Although some old-time pathologists remain skeptical, your lecturer believes that it will soon be standard practice to check all first- and second-degree relatives of a person (especially someone under age 18) who dies suddenly and has a negative autopsy, or who has near-sudden-death. In a 2005 Dutch study of the families of under-40 sudden death victims, 17 of 43 had a gene (catecholaminergic polymorphic V-tach was most common, followed by long-QT, Brugada, arrhythmogenic right ventricular dysplasia, hypertrophic cardiomyopathy Circulation 112: 207, 2005). Check includes EKG, exercise testing, and echocardiography. In a 2007 series limited to youngsters who died, a hereditary disease was found in 14 of 25 families (Pediatrics 120: 3967, 2007). European pathologists suggest professional standards for medical examiners inclding molecular examination of "negative autopsies" in the young (For. Sci. Int. 156: 138, 2006; I don't predict this will become standard in today's cost-conscious USA medical examiners' offices.) Diagnosis before birth, with confirmation by genetic studies after birth: Circulation 128: 2183, 2013 -- no one knows how many stillbirth are caused by channelopathies.
You also know that prolongation of the QT interval in response to antipsychotic drugs predicts sudden unexpected death from drug-induced rhythm disturbance: Lancet 355: 1048, 2000.
 |
Sudden ventricular fibrillation with no anatomic findings at autopsy.
Previous EKG's showed:
Usually men, more common among people of Asian ancestry, usually die in their sleep at night. Runs in families of course, and worth getting an implanted defibrillator for if you have it (Am. J. Card. 83: 98-D, 1999); there's no way to tell which Brugada patient is at risk of sudden death (J. Am. Coll. Card. 57: 2340, 2011).
About 3% of Thai and Laotian males have Brugada, and this accounts for the flap about "delayed death from yellow rain in Laotian immigrants" in the 1980's.
The sodium channel mutation (LQT3 / SCN5A, "idiopathic ventricular fibrillation" -- Nature 392: 293, 1998) is the usual Brugada gene.
* Less deadly, not Brugada, but interesting to scientists: KVLQT1 is a gene for familial atrial fibrillation (Science 299: 251, 2003).
* "Catecholamine-sensitive / catecholaminergic polymorphic V-tach" is a childhood disease in which exercise reproducibly causes the rhythm disturbances. As I predicted, it's a channelopathy, around half of cases it's in RYR2 (Mayo Clin. Proc. 79: 1367, 2004.
* "Idiopathic ventricular fibrillation", not familial and not caused by stress, surely covers a few entities that have not yet been discovered. Update on this and other causes of sudden cardiac death in structurally normal hearts: J. Am. Coll. Card. 43: 1137, 2004. Don't let this make anyone a cardiac neurotic, but "early polarization", a supposedly-harmless variant seen in maybe 5% of adults, is seen among 31% of people with "idiopathic V-fib" (NEJM 358: 2016, 2008).
* Myotonic dystrophy with abnormal EKG? NEJM 358: 2698, 2008
NOTE: Syncope can warn of most of these. How?
NOTE: The other major causes of sudden unexpected natural death are pulmonary emboli, pulmonary hypertension (idiopathic or in chronic lung disease), anaphylaxis, brain hemorrhages, and epileptic seizures ("SUDEP"; "Jett Travolta's disease"; Lancet 378: 2028, 2011; Neurology 57: 430, 2001; Neurology 64: 1131, 2005; NEJM 365: 1801, 2011; future medical examiners see Am. J. For. Med. Path. 23: 307, 2002). Since epileptics are not observed simply to drop dead, and "SUDEP" cases all follow a witnessed seizure or are diagnosed when an epileptic is found dead, typically in bed, other names have been proposed. The pathophysiology must involve interference with respiration (airway obstruction by objects or the throat itself; seizure spreading to the respiratory brain centers).
OTHER PROBLEMS IN CARDIAC ISCHEMIA
There's no room here to talk about the various rhythm disturbances and kinds of heart block that may result from coronary insufficiency.
Worth remembering: ATRIAL FIBRILLATION is a troublesome rhythm disturbance affecting about 3 million people in the US, seen in coronary disease, mitral valve disease (why?), hyperthyroidism (George Bush Sr.; why?), ectopic (and ablatable) foci in the pulmonary veins (NEJM 339: 659, 1998; now mainstream NEJM 354: 934, 2006), etc. It is especially dangerous because thrombi tend to form in the quivering atria and embolize; this causes 1/3 of strokes in older folks. (* Hereditary a-fib: NEJM 336: 905, 1997). For the hard-cases, the left atrial appendage occlusion device (to prevent embolization) is now fast-tracked for development (NEJM 360: 2601, 2009).
Serious degrees of HEART BLOCK can cause people with coronary disease (or other problems; remember amyloid) to drop over suddenly ("Stokes-Adams attacks", etc.) Get them pacemakers.
Despite the slight statistical advantage of a one-drink-a-day person over a non-drinker with the same other risk factors (Br. Med. J. 312: 1200, 1996; Br. Med. J. 314: 18, 1997), "your heart" is no reason to drink if you'd prefer not to. Because of the media hype over red wine ("tannins in wine protect the heart, the French paradox"), a couple of groups have looked at the cardioprotective properties of how-much vs. what-kind of alcohol (Br. Med. J. 312: 731 & 736, 1996); it's how much alcohol, not the kind of beverage.
It's now painfully clear that a person's successful return to near-normal living after a heart attack is largely a function of his or her knowledge about the disease. Fatalism turns you into a cardiac neurotic, a bad way to live. Education is the key to success. The implications for a primary-care physician are clear (Br. Med. J. 312: 1191, 1996).
No, it's probably not going to happen because you were having sex, even if you have angina or had a myocardial infarct (JAMA 275: 1405, 1996). Well... if you are reasonably fit, you are probably not going to have a heart attack during sex, but if you're a heart patient and out of shape and then go exercise and/or have sex, you're in danger (JAMA 305: 1225, 2011). This pathologist thinks there is no reason to tell people who have had heart attacks not to do what they want.
HIBERNATING MYOCARDIUM: "Chronic but reversible ischemic dysfunction". The most interesting discovery in "heart pathology" in recent memory (NEJM 339, 173, 1998; update Am. J. Path. 160: 1425, 2002).
This seems to be an adaptation that allows fairly good survival despite poor oxygenation (Am. J. Card. 98: 1574, 2006). Pathologists can spot hibernating cells (at autopsy, of course) by...
* You can usually find a few of cells with the same morphology normally in the subendocardium.
A similar subject is the "myocardial stunning" seen after heart surgery; the cause is apoptosis of cells (Ann. Thor. Surg. 73: 1229, 2002).
|
|
|
HYPERTENSIVE HEART DISEASE
In systemic hypertension, the left ventricle undergoes hypertrophy and, later, dilatation.
Probably the hypertrophy is mostly the result of pushing against the greatly increased load (more blood, more vascular resistance). Some hypertensives do have abnormally high cardiac outputs, so perhaps in these folks the heart is over-working for its own reasons.
There are rumors that some hypertensives suffer from chronic excess of catecholamines, and perhaps this exacerbates the hypertrophy.
Ultimately, the hypertensive's left ventricle will probably fail. To make the diagnosis, the heart must weigh more than 350 gm, and the left ventricle be more than 1.5 cm thick, with no other reason. You remember the microscopic appearance of the hypertrophied myocardial cell (thick fibers, many-ploid squared "boxcar nuclei"). It helps if the patient has a history of "high blood pressure", but when congestive heart failure supervenes on the salt-overloaded, hyper-constricted vascular system, "the high blood pressure may be cured".
NOTE: We talk about the etiologies of systemic hypertension under "Kidney".
* NOTE: Somebody may tell you that most congestive heart failure in the elderly is due to coronary atherosclerosis, even in the absence of ischemic scarring, and that the ischemia causes the hypertrophy. This pathologist doesn't follow this logic. If that were true, then probably you could build skeletal muscle by holding your breath.
Remember that hypertension is also an important risk factor for atherosclerosis, stroke, and so forth.
COR PULMONALE
A quaint name for a very serious problem. Any right-heart problem resulting from increased pulmonary vascular resistance (usually poor ventilation, less often fibrosis or primary vascular problems).
The cause of the poor ventilation may be anything from emphysema to a kyphoscoliosis. (If the emphysema patient is a "blue bloater" with extra-low oxygen in the alveoli, the lung vessels will of course constrict even more. However, you'll see plenty of pink puffers with cor pulmonale.)
"Cor pulmonale" would also include right-sided heart extra burden from narrowing of the pulmonary vascular tree (idiopathic pulmonary hypertension, Wegener's, pulmonary plexiform angiopathies, etc.) Whether "cor pulmonale" includes failure due to pulmonary emboli is a question for semantics experts.
You already know how pulmonary ventilation causes increased pulmonary vascular resistance ("causes pulmonary hypertension"). The right ventricle undergoes hypertrophy ("Feel that sternal heave!"), dilates, loses its familiar crescent shape and becomes more rounded, and eventually fails.
The polycythemia that accompanies hypoxia make the blood more viscous and prone to clot. This doesn't make the heart's job easier.
Increased pulmonary vascular resistance is a great impediment to a person's well-being. Often the right ventricle's problems set the real limit on quality of life in lung disease.
NOTE: The strained right ventricle is extremely vulnerable to rhythm disturbances. Patients with emphysema and cor pulmonale typically die very suddenly and unexpectedly as a result of an electrical storm in their strained ventricle. This mechanism probably underlies many (if not most) deaths from pulmonary emboli.
CONGENITAL HEART DISEASE: INTRODUCTION
|
|
|
|
|
Around 6-8 babies out of every 1000 has some kind of significant cardiac malformation.
Worth remembering:
Most of the time, nobody really knows why one heart forms normally and the next one doesn't (even folks who study the molecular biology have no breakthroughs: Ped. Clin. NA 53: 989, 2006). If one sibling has a defect, the next sibling has a 5% chance of having some (not necessarily the same) defect. Both monozygotic and dizygotic twins have a someone increased risk (Circulation 128: 1182, 2013).
Around 5% of cardiac defects are attributed to the chromosomal abnormalities, and the others now account for <1% of new cases.
Classic "SIDS" cases (i.e., the child was reported to be asleep) seldom reveal a cardiac malformation, but in the much less common situation in which a child falls dead while awake is often caused by a cardiac anomaly (and remember long QT): J. Ped. 141: 336, 2002.
A "shunt", of course, is abnormal flow of blood from one part of the circulation to another, which aren't supposed to communicate directly. Intracardiac shunts are usually the result of birth defects.
Remember that intra-cardiac shunts that produce turbulence are also prone to get infected (bacterial endocarditis; why?) As with other lesions with abnormal flow, look for JET LESIONS where turbulence leads to endocardial thickening, and sometimes fibrin deposition and even infection.
CONGENITAL HEART DISEASE WITH RIGHT-TO-LEFT SHUNTS ("early cyanosis"; "blue baby", "cyanotic congenital heart disease")
"Cyanosis", you remember, refers to a concentration of >=5 gm/dL of unoxygenated hemoglobin in the arterial blood. Kids who are chronically hypoxic as a result of right-to-left shunts may become polycythemic. If the hematocrit gets about 65% or so, the blood becomes over-viscous and hard to pump. Polycythemic blood also clots more readily (why?)
People with right-to-left shunts develop clubbing of the digits.
* The very cyanotic baby who requires urgent repair is likely to have serious brain damage from the time before surgery: Neurology 81: 241, 2013. This is visible both on imaging and clinically when these children are teens (J. Thorac. Card. Surg. 146: 543, 2013 -- D-transposition survivors).
You also know that these kids are prone to paradoxical embolization (i.e., a systemic venous embolus goes directly to the systemic arterial circulation), and particularly brain abscesses due to septic emboli.
* Not surprisingly, we now know that the small coronary arteries are substantially wider in these patients than in normal folks, with more blood being delivered to the heart muscle. We must think this is an adaptive change, though the mechanism remains unknown (Circulation 114: 196, 2006).
* Less healthy -- since the pulmonary arteries don't get the expected amount of blood under the expected pressure, they don't develop normally. The histopathology includes fragmented elastic, too much ground substance, and disarry of smooth muscle (Ann. Thorac. Surg. 87: 589, 2009).
Tetralogy of Fallot (Lancet 374: 1462, 2009).
Sketch it, and it makes sense. In the usual model, these babies have:
1. Overriding aorta (i.e., it straddles the ventricular septum, which is possible only because it has a hole in its top)
2. Pulmonic stenosis (usually, there is both stenosis of the pulmonic valve, which is often two-cusped, and much hypertrophy of the muscle trabeculae surrounding the track out of the right ventricle toward the pulmonary artery). This is the main determinant of severity in individual cases.
3. Ventricular septal defect (of course)
4. Right ventricular hypertrophy (obviously, since it must work so hard to perfuse the lungs through the defective pulmonic valve).
* If there is also an atrial septal defect, it is of course "pentalogy of Fallot". Around a quarter of patients have a right-sided aortic arch.
As you'd expect, these babies have a right-to-left shunt, and are born cyanotic. The lungs are hypoperfused (of course), and therefore won't suffer the ill-effects of lung hyperperfusion seen in left-to-right shunting.
The narrowed right-ventricular outflow track is the usual site of endocarditis.
"Tet attacks" are cyanotic episodes triggered by anything that briefly triggers increased pulmonary vascular resistance. A minor mystery of medicine is why these patients squat in the knee-chest position to relieve this. One investigator found that squatting changes the aortic wave form (Am. J. Hypertens. 15: 986, 2002); I have wondered if the altered shape of the thoracic cavity is sufficient to push the septum upward and partially close the VSD. Nobody knows.
Most of these people benefit from surgery.
Complete repair, with careful dissection of the pulmonary outflow tract and repair of the ventricular septal defect, was not available until the 1950's; it has become more and more successful. Mild cases of tetralogy may present in adulthood and most of these people do well after operation (Ann. Thorac. Surg. 83: 1790, 2007).
Histologic study of the resected muscle, mostly from the resected crista supraventriculis, often shows tremendous hypertrophy, and usually much interstitial fibrosis and endocardial thickening (J. Thor. Card. Surg. 132: 270, 2006). Not surprisingly, the little pulmonic trunks also show a variety of histologic abnormalities (J. Am. Coll. Card. 54: 1883, 2009).
{49021} tetralogy of Fallot
{08627} tetralogy of Fallot
{08630} tetralogy of Fallot
{08483} clubbing
* TAUSSIG-BING: "Kind of the opposite of Tetralogy of Fallot", with the aorta arising from the right ventricle and the pulmonary artery overriding the ventricle ("double outlet right ventricle"), which in Taussig-Bing bears a subpulmonic ventricular septal defect.
TRANSPOSITION OF THE GREAT ARTERIES (or "great vessels"): A family of disorders in which the aorta arises from the anatomic right ventricle, the pulmonary artery from the anatomic left ventricle. Usually the aorta is anterior and to the right of the pulmonary artery at their origin.
In UNCORRECTED TRANSPOSITION ("dextro-"), the blood flow is:
right atrium --> right ventricle --> aorta
left atrium --> left ventricle --> pulmonary artery
If the child is to survive for any length of time after birth, an atrial septal defect (usually septum primum) is almost always present; around 1/2 have a ventricular septal defect and 1/3 a patent ductus. Surgeons may be able to switch the aorta and pulmonary artery for a cure, or switch the atria (Mustard and Senning operations). There are now many survivors, and many do very well into adulthood (Circulation 114: 2699, 2006).
In CONGENITALLY CORRECTED TRANSPOSITION ("levo-"), in adition, the atria are switched relative to the ventricles by the malformation, so that the blood flow is:
right atrium --> left ventricle --> pulmonary artery
left atrium --> right ventricle --> aorta
This wouldn't be a problem, except that with this setup, the tricuspid and mitral valves can't really be normal. This may present in adulthood and also benefit from surgery.
{08644} transposition of the great vessels
{08647} transposition of the great vessels
{08650} transposition of the great vessels
{40920} transposition of the great vessels
![]() Transposition of the great vessels
Transposition of the great vessels
Pittsburgh Pathology Cases
* There are many other variants. In DOUBLE-OUTLET RIGHT VENTRICLE, both pulmonary artery and aorta arise from the right ventricle.
* We are just now beginning to study the brain abnormalities in these children, which seem to result from the poor perfusion during development (NEJM 357: 1928, 2007).
TRUNCUS ARTERIOSUS
The pulmonary artery and aorta are a single vessel, overriding a ventricular septal defect. Their valve typically has four cusps, etc.
Eventually the lungs are damaged by the high-pressure flow into their arteries.
* Concurrent defects of the atrioventricular septum are common; for some reason, when this happens the trunk arises just from the right ventricle. (Ann. Thorac. Surg. 87: 1495, 2009).
{08656} truncus arteriosus
TRICUSPID ATRESIA
"Atresia", of course, means failure of a normal lumen to develop.
Obviously, survival is only possible if there is an atrial septal defect and some other route to get the blood to the lungs. (Think! How could this happen?)
The prognosis is still guarded. Closure of the ductus during the first few days of life can be a catastrophe (why?)
"Ebstein's anomaly" is a curious lesion with a low-slung valve outlet, large anterior leaflet and functional problems that vary in severity from patient to patient. The right atrium may be huge and the right ventricle tiny. As you'd expect, there are often major problems filling the right ventricle and/or serious tricuspid regurgitation; often there is an atrial septal defect and/or some other problem involving the left side as well (Mayo Clin. Proc. 80: 361, 2005). Surgery may involve creating a single ventricle, though today the two-ventricle approach is favored by some groups (Ann. Thorac. Surg. 84: 587, 2007). Valvuloplasty: Ann. Thor. Surg. 83: 676, 2007. Today's options: Ann. Thorac. Surg. 96: 1703, 2013.
HYPOPLASTIC LEFT HEART (Autopsy series J. Thor. Cardio. Surg. 144: 1301, 2012.
Underdevelopment of the aortic and mitral valves, left ventricle, and proximal aorta. The blood returning from the lungs goes through the atrial septal defect.
Severity ranges from mild hypoplasia to atresia of the mitral and aortic valves without any left ventricle at all. Altogether, these are quit common, around 2.3 per 10,000 live births (J. Am. Coll. Card. 39: 1890, 2002).
Untreated, this is lethal shortly after birth, though compatible with normal life as an unborn child while the ductus stays open.
Today's three-stage procedure works like this.
* One of the "great ethical debates" of the early 1990's led to the prohibition
of the use of anencephalic babies' ("Baby Theresa") hearts to treat hypoplastic left heart syndrome.
These used to save lots of lives.
After "Baby Theresa", about half of these kids ended up dying as a result of the hoopla
(NEJM 330: 501, 1994).
*"Baby Fae" was born with a hypoplastic left heart.
She was given a baboon's heart by a surgeon interested in "independent
scientific thought".
Do you remember the press conference in which the surgeon predicted she'd live
a normal life? Most people would consider the whole business unethical.
I think that reasonable people can disagree about this.
Please remember this was the first infant heart transplant, and prepared
the way to many lives saved, especially from anencephalic donors before
saving lives in this way was made illegal.
{11483} hypoplastic left heart
|
* Vivien Thomas, the famous surgical genius who was assistant to and collaborator with Alfred Blalock, was an African-American who because of the racism of his era never obtained formal education beyond high school. While developing operative procedures for tetralogy and other birth defects at Vanderbilt in the 1930's, he had to be classified and paid as a janitor. As recognition of his part in the development of "blue baby surgery", one of the colleges in Johns Hopkins medical school is named for him. His autobiography is "Partners of the Heart." See JAMA 300: 328, 2008.
CONGENITAL HEART DISEASE WITH LEFT-TO-RIGHT SHUNTS ("late cyanosis" / "tardive cyanosis")
A family of diseases in which much of the left cardiac output returns to the pulmonary system. This eventually causes serious damage to the pulmonary vasculature.
This is bad. The left-to-right shunt will make the heart work harder, and the increased blood flow to the lungs will eventually damage the pulmonary arteries. When pulmonary artery pressure exceeds aortic pressure, the shunt will reverse ("Eisenmenger's syndrome"), cyanosis will occur, and death will follow shortly.
Eisenmenger's is a major disaster. Long-term prostacyclin (PGI2) for these people: Circulation 99: 1858, 1999.
Circulating endothelial cells as a marker for irreversibility of pulmonary arterial damage in left-to-right shunting: Circulation 119: 374, 2009.
VENTRICULAR SEPTAL DEFECT ("VSD", "Roger's disease" if <=5 mm, etc.): A hole in the ventricular septum. This can be serious or trivial (Lancet 377: 1103, 2011).
90% of the VSD's you'll pick up in older children and adults are in the membranous septum. Unfortunately, this is near the bundle of His; these patients may develop heart block.
The remaining 10% of clinically significant VSD's are in the muscular septum. (In newborns, the majority of VSD's are in the muscle, but most of these close, so they don't count.)
The best thing about VSD is that around 50% close spontaneously, either when the muscle hypertrophies or the tricuspid leaflet adheres to the defect. Natural history: Arch. Dis. Child. 81: 413, 1999.
When treatment is necessary, some can be closed by percutaneous endovascular procedures with placement of little VSD occluders. Larger defects still require open-chest surgery.
{03281} ventricular septal defect
{03287} ventricular septal defect (sorry about ruler over hole)
{03635} membranous ventricular septal defect
{03638} membranous ventricular septal defect
{08525} ventricular septal defect
{03650} complete atrioventricular septum defect
|
|
|
|
The most extreme example is "univentricular heart" (Circulation 115: 800, 2007) with no septum at all. (When there is a good atrial septum, it is called "cor triloculare biatrium", Why the name?)
* Surprisingly, some of these these patients can do remarkably well without surgery (Am. J. Card. 77: 542, 1996).
ATRIAL SEPTAL DEFECT: A hole in the atrial septum.
Among these:
90% are OSTIUM SECUNDUM DEFECTS, problems with the foramen ovale ("patent foramen ovale", etc.). Often this is just a probe-patent slit where the flap joins the rest of the muscle. If a flap covers such a defect, it may re-open in the setting of pulmonary hypertension (i.e., pulmonary embolization) and explain paradoxical embolization.
Where there's a real, obvious hole, there will always be some right-to-left shunting whenever central venous pressure exceeds left atrial pressure. Hence the danger of paradoxical embolization.
Many people (estimates vary widely) have a probe-patent foramen ovale (i.e., the autopsy pathologist can slide a probe through the atrium by twisting it around the membrane). In these people, there's a clear link to cryptogenic stroke (Neurology 81: 619, 2013), and of course it's a possible danger for scuba divers (South. Med. J. 99: 1367, 2006). There is a current claim that closing this transcutaneously is an effective treatment for migraine; the procedure seems safe enough and perhaps there'll be some hard science soon (Neurology 65: 1415, 2005; Chest 130: 896, 2006; Am. J. Card. 99: 1316, 2007; Nat. Clin. Pract. Cardio 3: 446, 2006). The latest controlled studies are disappointing but I was impressed with anecdotes.
* Early surgical closure to prevent rhythm problems after the operation: NEJM 348: 839, 1999.
Of course, with echo we're picking up lots more kids with secundum defects; the majority, especially the smaller ones, tend to close spontaneously (Pediatrics 118: 1560, 2006 -- I wonder if these perhaps re-open if pulmonary hypertension develops).
Young people presenting with stroke for no obvious reason (and especially no terrible risk factors for atherosclerosis) tend to have patent foramen ovale (Heart 98: 485, 2012). Your lecturer is wondering whether everybody may get screened. Catheter procedures are now the norm with only the largest atrial septal defects requiring surgical closure. Repairing patent foramen ovale using devices is now routine and very effective in improving these folks' physiology: Am. J. Card. 112: 1258, 2013.
5% are SINUS VENOSUS DEFECTS, near the superior vena cava. These people are likely to have one or more of the right pulmonary veins that return to the right atrium or superior vena cava, rather than the left side.
5% are OSTIUM PRIMUM DEFECTS, low on the septum at the cardiac crux. Often these are Down's children, and there's generally some mitral insufficiency, too.
* "Coronary sinus type" is rare. Look for a persistent left superior vena cava.
* LUTEMBACHER'S SYNDROME merely refers to mitral stenosis plus an atrial septal defect. What do you think happens?
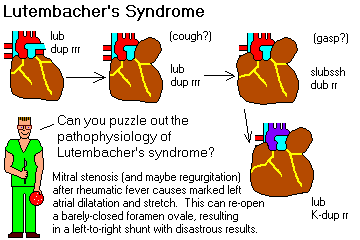
Functionally significant atrial septal defects need surgery or transcatheter repair to prevent the same pulmonary problems seen in VSD's, as well as the often-devastating effects of a right-to-left shunt.
{08498} atrial septal defect
{08501} atrial septal defect
{08513} atrial septal defect
PATENT DUCTUS ARTERIOSUS
The ductus ordinarily closes in the first few days of life. Prostaglandin E apparently keeps it open, and closure can be induced in many cases using prostaglandin antagonists.
If the ductus fails to close, the child's "machinery murmur" needs to be turned off by surgical ligation. If not, the same problems with left-to-right shunting seen with VSD and ASD will occur.
Note that closure can exacerbate certain cardiac birth defects. In pulmonic stenosis, transposition of the great vessels, and hypoplastic right heart syndrome, it's best for the ductus to remain open to supply blood to the lungs. More about this on rotations.
* Patent ductus, left open, is prone to get infected with bacteria.
{08566} patent ductus arteriosus
{08569} patent ductus arteriosus
VARIATIONS
"Endocardial cushion defects" / "complete atrioventricular septal defects" / "common AV canal" are a group of defects that involve the formation of the AV valves, the anterior atrial septum, and the posterior part of the ventricular septum.
{08603} AV canal
{08617} AV canal
OBSTRUCTIVE, NON-CYANOTIC CONGENITAL ANOMALIES ("acyanotic")
COARCTATION OF THE AORTA
A fibrous ring around a portion of the aorta, or perhaps another large artery. This is common in Turner's, or may be idiopathic.
If the occlusion is pre-ductal (the rare "infantile type", i.e., proximal to the ductus arteriosus), patents may even have heart failure while in the womb. In this case, the upper half of the body is supplied by the aorta, and the lower half is supplied by the pulmonary artery via a patent ductus arteriosus.
In the post-ductal (common, "adult") type, the stenosis is distal to the ductus arteriosus. There's a considerable pressure gradient along the narrowed segment. Since the lower half of the body is likely to be under-perfused (claudication, etc.), renal hypertension is usual, and collateral arterial circulation will develop (via which arteries? HINT: It notches the ribs!) If the femoral pulses on a hypertensive patient seem late and weak, it's probably coarctation of the aorta.
You want to get most cases of "co-arc" fixed, not the least reason being that the site is prone to become infected. Surgical and catheter-based options are both in widespread use (J. Am. Coll. Card. 43: 1062, 2004; Ann. Thorac. Surg. 80: 1659, 2005; AJR 187: W302, 2006).
Even after repair, hypertension can be a problem, especially when exercising (underperfusion of the kidneys: Am. J. Card. 99: 1284, 2007).
PULMONARY STENOSIS / STRESIA WITH INTACT INTERVENTRICULAR SEPTUM
Not rare. Obviously, the blood must flow to the left side of the heart (usually through an atrial septal defect), and then to the lungs via a patent ductus.
This is a very serious lesion, and prognosis depends largely upon how much of the right side of the heart has formed properly, and whether the coronary circulation might be compromised by decompression of the right ventricle by the shunt that will need to be placed (for example, there's often a fistula between a coronary and the right ventricle). See Ann. Thor. Surg. 84: 574, 2007.
Today, the diagnosis may be made before birth, and a catheter introduced to try to improve the opening. Details Am. J. Card. 99: 699, 2007.
AORTIC VALVE STENOSIS AND ATRESIA (review of decades of autopsy work Am. J. Card. 112: 541, 2013)
In congenital VALVULAR aortic stenosis, the valve is a fibrous diaphragm.
In congenital SUB-VALVULAR aortic stenosis, there is a thick, fibrous ring below the valve itself.
In congenital SUPRA-VALVULAR aortic stenosis, there's a similar ring just past the valve. * Thankfully rare. This one's genetic. One mutation is now known to be the elastin gene: Hum. Genet. 106: 577, 2000, Am. J. Card. 83: 1141, 1999, others.
| Milder birth defects of the aortic valve are much more common. BICUSPID AORTIC VALVE, seen in around 1% of folks, produces more "congenital heart disease" patients with real problems than any other among the birth defects. It tends to calcify later in life, and become stenotic. The right coronary and left coronary leaflets are the most likely to be fused; patients with the non-coronary cusp fused to the right coronary cust are more likely to have trouble (J. Am. Coll. Card. 44: 1648, 2004). Pathologists see J. Thor. Card. Surg. 133: 1226, 2007 for a subclassification based on number and position of raphes and whether there is stenosis; at the very least, report the number of raphes, if any. There may be a small increase in risk for aortic dissection (JAMA 306: 1104, 2011). | 
|
UNICUSPID AORTIC VALVE, a rarity, may also remain asymptomatic. Tetracuspid valve is a curiosity.
{06779} bicuspid aortic valve
{46205} bicuspid aortic valve
{06818} bicuspid aortic valve, this had both stenosis and insufficiency
{06785} unicommissural aortic valve
{53319} aortic valve with four cusps
![]() Bicuspid aortic valve
Bicuspid aortic valve
WebPath photo
* In the 1990's, there was a flap over
chlamydia![]() as contributing to the calcification of normal aortic valves,
just as they were once blamed for atherosclerosis. I predicted
in 1996 that this will prove to be non-related, and that the bugs
were just hangers-on. This now seems to be correct
(Am. J.
Coll. Card. 29: 1054, 1997;
Circulation 99: 2733, 1999; Mayo's Atherosclerosis 174:
337, 2004) though some groups still find a relationship
(?? immune complexes Eur. Heart J. 24: 198, 2003).
as contributing to the calcification of normal aortic valves,
just as they were once blamed for atherosclerosis. I predicted
in 1996 that this will prove to be non-related, and that the bugs
were just hangers-on. This now seems to be correct
(Am. J.
Coll. Card. 29: 1054, 1997;
Circulation 99: 2733, 1999; Mayo's Atherosclerosis 174:
337, 2004) though some groups still find a relationship
(?? immune complexes Eur. Heart J. 24: 198, 2003).
MALPOSITIONS OF THE HEART
An ACARDIUS is a birth defect in which there is no heart. If the pregnancy goes to term, it is always because the child shares circulation with a twin.
{21109} acardius
{21110} acardius
DEXTROCARDIA means the heart's on the right side. In Kartagener's syndrome, the cilia are immotile due to lack of dynein arms, so the organ often end up backwards. In SITUS INVERSUS TOTALIS, with everything backwards, the heart is usually well-formed. If the heart is the only organ that is mal-positioned, it often bears other defects.
ENDOCARDIAL AND VALVULAR DISEASE
The page [Lydgate] opened on was under the head[ing] of Anatomy, and the first passage that drew his eyes was on the valves of the heart. He was not much acquainted with valves of any sort, but he knew that valvae were folding-doors, and through this crevice came a sudden light startling him with his first vivid notion of finely adjusted mechanism in the human frame... The moment of vocation had come, and before he got down from his chair, the world was made new to him... From that hour Lydgate felt the growth of an intellectual passion.
-- George Eliot, "Middlemarch"
|
|
|
|
|
|
|
|
|
|
|
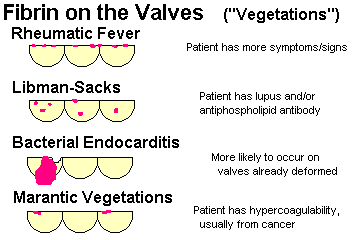
KNOW YOUR COMMON ETIOLOGIES!
MITRAL STENOSIS (Lancet 374: 1271, 2009)
Old rheumatic fever
We are now seeing occasional older folks with severe calcification of the mitral valve annulus who get some mitral valve stenosis. No one knows what to do about this.
(All other causes are very uncommon -- Libman-Sacks is perhaps "the other cause"; see Am. J. Med. 120: 636, 2007. Fabry's and some of the mucopolysaccharidoses are occasionally causes)
NOTE: Isolated mitral stenosis is pretty well tolerated most of the time.
MITRAL REGURGITATION (Lancet 373: 1382, 2009)
Old rheumatic fever
Bacterial endocarditis
Barlow's syndrome / degenerative mitral valve disease
Other birth defects at the crux
Ruptured papillary muscle (MI)
Ruptured chorda (bacterial endocarditis, Barlow's)
Dilated annulus (left CHF)
Libman-Sacks
Calcified mitral annulus (maybe)
Mucopolysaccharidosis (why? -- Hunter's Ann. Thor. Surg. 80: 1911, 2005).
Degenerative changes of old age (the most common today: NEJM 335: 1417, 1996)
Ischemic myocardial disease or cardiomyopathy, with the valve stretched out of shape -- now a surgical disease in itself (J. Thorac. Card. Surg. 142: 995, 2011).
AORTIC VALVE STENOSIS (Lancet 373: 956, 2009)
Old rheumatic fever
Congenital bicuspid valve that calcified
Normal valve that calcified
Birth defects (valvular, sub-valvular)
Libman-Sacks
Alkaptonuria ("somebody poured India Ink all over the aortic valve")
NOTE: Some of the biggest hearts in clinical medicine result from aortic valve stenosis. Obviously, the pulse pressure is narrowed, and systole is prolonged. This can have very bad consequences for myocardial perfusion.
NOTE: As I trust you've figured out, "aortic stenosis" (by custom) refers to stenosis of the valve, not the aorta. If a portion of the aorta is stenotic, it's called "coarctation". If the entire aorta is stenotic, you didn't get born.
AORTIC REGURGITATION (review Circulation 99: 1851, 1999 urges aggressive surgical management)
Old rheumatic fever
Bacterial endocarditis
Syphilis![]()
Dissecting hematoma / steering wheel injury
Marfan's
Libman-Sacks
Fenfluramine (diet pill fiasco): about 9% of those treated got aortic regurgitation on echo, but cardiac symptoms are not much greater than in controls: JAMA 283: 1703, 2000.
Behcet's (can produce an endocarditis: Medicine 91: 25, 2012)
The ring dilates:
Rheumatoid arthritis
Ankylosing spondylitis / HLA-B27 family
Syphilis![]()
"Annuloaortic ectasia" (usually older hypertensives)
A few patients with VSD's have a leaflet prolapse into the defect
NOTE: You'll learn about increased pulse pressure, "Corrigan's jumping pulse", pistol-shot sign, etc., etc. on rotations. The heaviest hearts in medicine have classically been "cor bovinum" in aortic regurgitation, sometimes over 1000 gm. The top weight in my series was 950 gm in morbid obesity, same as in Am. Heart. J. 130: 306, 1995.
TRICUSPID STENOSIS
Old rheumatic fever
Carcinoid heart disease (usually this gives tricuspid insufficiency instead)
TRICUSPID REGURGITATION
Old rheumatic fever
Carcinoid heart disease
Bacterial endocarditis (ask about IV drug use!)
Loeffler's
Dilated annulus (right CHF)
Libman-Sacks
NOTE: Look for those jumping neck veins!
PULMONIC STENOSIS
Tetralogy of Fallot
Congenital ("funnel")
Carcinoid heart disease
PULMONIC INSUFFICIENCY
Dilated annulus (right CHF). Rare.
Carcinoid heart disease (usually this gives pulmonic stenosis instead)
KNOW YOUR VEGETATIONS!
ACUTE RHEUMATIC FEVER
Small, warty, sterile, on the lines of closure and perhaps chordae
Seldom embolize
BACTERIAL ENDOCARDITIS
Often large, loaded with bacteria
Find them on any deformed intracardiac surface
Very prone to embolize
NON-BACTERIAL THROMBOTIC ("MARANTIC") ENDOCARDITIS
Small, sterile, on the lines of closure
May embolize
LIBMAN-SACKS ENDOCARDITIS -- lupus and antiphospholipid antibody syndrome
Usually 4 mm or less, sterile, on either surface of the leaflet / chordae
May embolize
NOTES: (1) You'll learn your murmurs in clinical medicine. "Is this a worrisome systolic murmur?" JAMA 277: 564, 1997. (2) As with any intracardiac lesion involving turbulence, deformed valves are prone to develop bacterial endocarditis. (3) Future pathologists: Again, look for jet lesions on the endocardium if you suspect a valve of having produced abnormal flow.
CALCIFIC AORTIC VALVE STENOSIS (BMJ 336: 550, 2008)
While bicuspid valves are notorious for calcifying later in life, sometimes a normal, tricuspid valve accumulates calcium-rich excrescences in its cusps. We now know that 3-5% of older folks will have at least some aortic valve calcification. Maybe 1 older man in 100 will need to be treated; women are at somewhat lower risk.
The etiology remains obscure, though of course "inflammation, just like in atherosclerosis" is commonly cited. I was not surprised, though, by the complete failure of intensive cholesterol-lowering to affect the disease (NEJM 359: 1343, 2008). An allele at the lipoprotein A locus: NEJM 368: 503, 2012.
The calcification is an active process; the local cells pump out osteopontin, a bone-laying-down protein (Circulation 92: 2163, 1995).
These can interfere with the valve opening, and can block the coronary ostia. The latter is very serious.
{03560} calcified tricuspid aortic valve
{06461} calcified tricuspid aortic valve
DEGENERATIVE MITRAL VALVE DISEASE ("mitral valve prolapse", "Barlow's", "floppy mitral valve", "ballooning mitral valve", etc.)
In this semi-disease that affects around 2-3% of humankind, the mitral valve has an elongated posterior leaflet with some extra ground substance ("myxoid degeneration"). The leaflet looks like a parachute canopy, and billows up into the atrium.
Today, we distinguish "classic Barlow's", in which the posterior leaflet is thickened and the chordae are long, with the famous mid-systolic click and late murmur, and "non-Barlow degenerative mitral valve disease". There are sub-subclassifications but none of this is really helpful. In any case, the process progresses over decades and today surgical correction may eventually be required (NEJM 363: 156, 2010).
The other valves may show the same thing. The histopathology exhibits weird derangements of all connective-tissue components (Am. Heart J. 129: 1149, 1995).
The typical patient has a slim build (the more "Marfanoid" you are, the most likely you are to have "Barlow's"), a mid-systolic click (the leaflet snapping tight) and ever-so-slight mitral regurgitation. The timing will vary with how full the ventricle gets.
Trivial as it is, mitral valve prolapse is the most common structural cause for people who present with palpitations. Maybe this has something to do with re-entry around scar in the mitral valve -- nobody really knows. Or maybe we look harder for "Barlow's" in people who just happen to have palpitations. Your lecturer is undecided.
There's differing opinions as to whether Barlow folks should take endocarditis prophylaxis prior to visiting the dentist.
If there's a stroke risk at all, it's small (Mayo Clin. Proc. 69: 632, 1994; NEJM 341: 48, 1999).
Occasionally, they get re-entry type atrial rhythm disturbances (notably "supraventricular tachycardia", in which one's heart "goes off like an alarm clock" at around 190 beats per minute, making adequate filling impossible. This is moderately annoying and happens to your lecturer, who has mild echo-proven Barlow's, if he neglects exercise for a while and thereby drops his vagal tone). There are rumors of people suddenly falling over dead with no abnormality except Barlow's. Is it causal? Who knows? It's very, very infrequent.
Occasionally a chorda will rupture, producing serious mitral regurgitation.
The floppy leaflets of the degenerating mitral valve are very suited for the new "clip", which buckles and tightens one or both leaflets and can be placed percutaneously with a result almost as good as open-heart procedures (NEJM 364: 1395, 2011).
The greatest Barlow morbidity in the young is probably that lots of them become "cardiac neurotics". This is really unfortunate. Natural history of Barlow's: NEJM 341: 48, 1999.
{06470} floppy mitral valve
{40363} floppy mitral valve
{03440} floppy aortic valve, from a Barlow's case
RHEUMATIC FEVER / RHEUMATIC HEART DISEASE (Lancet 379: 953, 2012)By and by, Beth said the needle was "so heavy", and put it down forever. Talking wearied her, faces troubled her, pain claimed her for its own, and her tranquil spirit was sorrowfully perturbed by the ills that vexed her feeble flesh. |

|
Rheumatic fever ("romantic fever"
etc., etc.) is a systemic, autoimmune disease
triggered by a beta-hemolytic
streptococcal![]() throat infection. (The major reason for going after "strep
throat" with penicillin is to prevent rheumatic fever; around 3-5% of untreated strep throat turns into
rheumatic fever.)
throat infection. (The major reason for going after "strep
throat" with penicillin is to prevent rheumatic fever; around 3-5% of untreated strep throat turns into
rheumatic fever.)
"Scarletina / scarlatina" is a red rash from a strep throat caused by a strep toxin; don't confuse it with RF.
The acute disease declares itself about two weeks after the "strep throat" gets better.
Most patients are kids 5-15 years old. There are about 300,000 to 350,000 cases each year, and 200,000 to 250,000 people die prematurely as a result of damage from the disease (Lancet 379: 953, 2012). In the developed world, it is mostly the poorer kids. (* HLA-B5 is a risk factor too; most recently, a susceptibility gene close to HLA has been predicted Circulation 89: 138, 1994) but nobody is immune. The disease is endemic in the poor nations (Ann. Int. Med. 120: 177, 1994; the Australian aboregine people Arch. Dis. Child. 95: 455, 2010 and Circulation 128: 492, 2013; "disadvantaged indiginous populations" Ped. Clin. N.A. 56: 1401, 2009; Africa's poor Circulation 120: 709, 2009), a poor area in Italy (J. Ped. 160: 832, 2012), and recently in resurgence in the U.S. (the West Pennsylvania outbreak, which featured a distinctive streptococcus subtype: J. Ped. 149: 58, 2006).
* Although the history and physical exam have been the usual way of old detecting rheumatic valvular disease, it's now clear that, at least in the poor nations, many patients without obvious physical exam findings (i.e., easy-to-hear mitral stenosis) have the pathology on echocardiography (NEJM 357: 470, 2007).
Update on the acute illness: Lancet 366: 155, 2005.
The classic teaching about the pathogenesis of the disease involves molecular mimicry. The M-protein of the streptococcus, and the more-recently-characterized G1NAc, are attacked by T-cells and antibodies which then cross-react with the heart (anti-myosin antibody: J. Inf. Dis. 168: 915, 1993, probably others), brain, skin, and joints.
* This can't be the whole story, of course, since some people have these antibodies without having rheumatic fever. Perhaps the streptococcal hyaluronidase damages the endothelium, or a hapten is involved, or....
The cardiac pathology is a pancarditis (i.e., any layer can be involved, even the pericardium). The characteristic lesion in the myocardium (especially near the vessels) is the ASCHOFF BODY (Aschoff nodule), a mass of fibrinoid surrounded by distinctive ANITSCHKOW CELLS ("caterpillar cells"), bloated, pale-purple-staining macrophages with a curious caterpillar-shaped mass of heterochromatin running the length of the nucleus (in cross-section, this is an "owl eye" instead). You may see big multinucleated forms, with large "eyes" in cross-section (ASCHOFF GIANT CELLS).
There's often a type III immune-injury type systemic vasculitis.
Curiously, textbooks differ about whether the Anitschkow cells are macrophages or altered myocardial cells. Their appearance is very different from macrophages anyplace else, and this observer thinks he sees transitional forms to nearby myocytes. However, they stain uncharacteristically for mature myocytes, but look like they bear immature mesenchymal nuclei (Pathology 31: 98, 1999). Stay tuned.
* Despite the textbooks, Aschoff nodules simply do not look like other granulomas, and I am not aware of any reason to think they should be considered granulomas.
Here are the symptoms and signs of rheumatic fever:
EVIDENCE OF CARDITIS (i.e., pericardial pain, enlarged heart, murmurs, failure)
POLYARTHRITIS
SYDENHAM'S CHOREA (St. Vitus's dance), from involvement of the basal ganglia. This is a big deal right now. Mediated by antibodies that cross-react with neurons (type II immune injury, non-destructive).
ERYTHEMA MARGINATUM, a snake-like red skin eruption
SUBCUTANEOUS NODULES, little masses of fibrinoid with granulomas around them, over the bony bumps on arms and legs.
FEVER
LAB EVIDENCE OF RECENT STREP INFECTIONS (i.e., a high anti-streptolysin O and/or anti-hyaluronidase and/or anti-DNAse titer, a culture result, or whatever)
Here are the JONES CRITERIA for making the diagnosis. You need one major and two minor, or two majors.
Major: Polyarthritis; carditis; chorea; subcutaneous nodules; erythema marginatum.
Minor: Arthralgia; fever; preceding group A strep infection; preceding bout of rheumatic fever; elevated sed rate; prolonged PR interval on EKG.
The inflamed heart may fail. More often, the endocarditis manifests as VERRUCAE, tiny sterile masses of fibrin and platelets along the lines of closure of the valve leaflets.
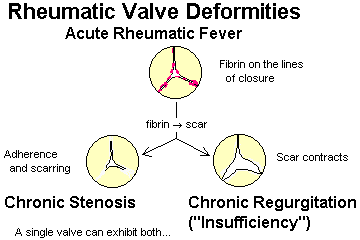
Unfortunately, the verrucae are prone to organize, causing deformity of the heart. If the cusps stick together, STENOSIS results. If scar contraction shortens and bends the valve leaflets, INSUFFICIENCY / REGURGITATION results.
The aortic and mitral valves are usually affected; the tricuspid sometimes, the pulmonic rarely.
Rheumatic fever usually ends in recovery after a few weeks, but recurrences are common and likely to be more severe.
Old rheumatic fever is one of the most common causes of cardiac valve disease.
If you suspect the cause of valve disease is rheumatic, look for FUSION OF THE CUSPS, where the verrucae have organized.
One hallmark of old rheumatic mitral valve disease is thickening of the chordae tendineae. Also, check the posterior aspect of the left atrium for the white, geographic fibrous patches called MACCALLUM'S PATCHES; probably these are just the jet lesions from mitral insufficiency.
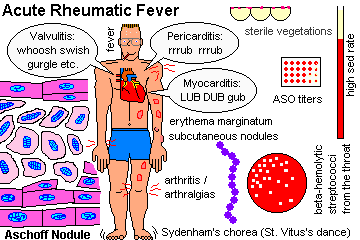
 {11489} old rheumatic fever, mitral valve
{11489} old rheumatic fever, mitral valve
{11492} acute rheumatic fever
{49008} acute rheumatic fever, vegetations
{18809} Aschoff nodule
{28587} Aschoff nodule
{28590} Aschoff nodule
{53304} Aschoff nodule
{46305} Aschoff nodule
{46481} Aschoff nodule, great caterpillar chromatin
{06467} old rheumatic fibrosis of mitral valve
{06845} old rheumatic mitral stenosis
{06847} old rheumatic mitral stenosis
{06850} old rheumatic mitral stenosis, large left atrium
{06815} old rheumatic aortic valve disease with insufficiency
{11549} old rheumatic aortic valve disease, good fusion of cusps
{11486} old rheumatic aortic valve stenosis
|
|
|
|
|
Australian Pathology Museum High-tech gross photos
|
![]() Mitral Stenosis (old rheumatic fever)
Mitral Stenosis (old rheumatic fever)
CDC
Wikimedia Commons
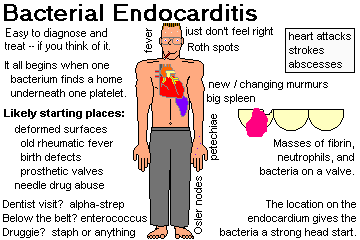
INFECTIVE (BACTERIAL) ENDOCARDITIS (Lancet 363: 139, 2004; Lancet 379: 965, 2012).
Blood flow in the normal heart is laminar. If the flow becomes turbulent (i.e., usually due to congenital or rheumatic deformities), endothelium is likely to be damaged.
If endothelium is damaged, tiny platelet-fibrin clots can form on the valves. This wouldn't be much of a problem, and they'd probably go away, except for that fact that every once in a while, a bacterium can get enwrapped in the clot.
Yes, there's often a bacterium or two in your bloodstream. If you brush your teeth or pass stool, you're going to get a few bacteria into your blood. If you go to the dentist or get catheterized, proctoscoped or cystoscoped, or have colon cancer, you'll get lots more. All of these place you at risk for endocarditis. And if you do IV drugs without using sterile technique, you're begging for trouble.
Once the bacterium has found a place to live under the fibrin cocoon, it is protected from the body's defenses, since the valve leaflets are avascular, and the blood in the chamber is flowing too fast to allow neutrophils to stop here. The surrounding endocardium will probably get damaged, and the vegetation can grow.
Sooner or later, the polys will notice the bacteria and start fighting them, but by this time, they are generally massive. They are going to cause problems.
Naturally, the foul breakdown products of these bacteria are going to make a person systemically sick. Fever, arthritis, the acute phase reaction, anemia of chronic disease, diffuse proliferative glomerulonephritis, evidence of B-cell hyper-activation (rheumatoid factor, cryoglobulins, false-positive syphilis test), clubbing, etc., etc., are prone to supervene.
The vegetations are notoriously crumbly ("friable"; why might this be? Hint: polys), and prone to embolize. Embolic phenomena include SPLINTER HEMORRHAGES of the nails (if they are in the proximal nailbed I'd worry), OSLER'S NODES (sore spots in the finger pulp), JANEWAY LESIONS (on the palms) and ROTH SPOTS (vasculitis where arterial branch-points have caught septic emboli, seen in the eyegrounds). More serious are infarcts (strokes, heart attack, and so forth) and the development of mycotic aneurysms (weakened arteries where a mass of bacteria has landed and caused damage) and/or abscesses throughout the body.
Bacterial endocarditis is rightly dreaded. Even nowadays, about 1 patient in 6 will die despite everything you can do (JAMA 288: 75, 2002). Autopsy series show the many complications that actually kill -- and remind us of the grim fact that bacterial endocarditis is still often missed clinically (100% fatal error) -- Medicine 91: 152, 2012.
* Future pathologists: Enjoy J. Inf. Dis. 193: 1711, 2006 for a technique to see the bacteria in a fatal case, even after treatment, using the patient's own tagged antibodies.
|
|
|
|
|
|
Australian Pathology Museum High-tech gross photos
|
|
|
|
|
|
|
|
|
![]() Bacterial endocarditis
Bacterial endocarditis
CDC
Wikimedia Commons
The vegetations (actually, probably the polys) are prone to eat through valve cusps and chordae, causing further damage and even perforation.
ACUTE BACTERIAL ENDOCARDITIS may affect a normal valve, is often caused by virulent
staphylococci![]() (less often, beta-hemolytic
streptococci
(less often, beta-hemolytic
streptococci![]() ,
gonococci, proteus, pseudomonas;), and is highly lethal
(death in less than 6 weeks). These people may be IV drug abusers or have a severe infection
elsewhere (especially if the infection is on the right side;
the higher pressures, greater turbulence, and higher oxygen concentrations
make the left side more popular with most bacteria). Or they have occult colon
cancer. Or they may just be unlucky.
,
gonococci, proteus, pseudomonas;), and is highly lethal
(death in less than 6 weeks). These people may be IV drug abusers or have a severe infection
elsewhere (especially if the infection is on the right side;
the higher pressures, greater turbulence, and higher oxygen concentrations
make the left side more popular with most bacteria). Or they have occult colon
cancer. Or they may just be unlucky.
SUBACUTE BACTERIAL ENDOCARDITIS generally affects cardiac surfaces where there's been turbulent blood
flow. The usual culprit is viridans α-hemolytic
streptococci![]() ; enterococci or
gram-negatives are
common if the person's had a below-the-belt medical procedure. Actually, lots of bacteria, as well as
candida
; enterococci or
gram-negatives are
common if the person's had a below-the-belt medical procedure. Actually, lots of bacteria, as well as
candida![]() and aspergillus
and aspergillus![]() (Arch. Path. Lab. Med. 129: 511, 2005), can produce subacute bacterial endocarditis.
The HACEK group (Hemophilus aphrophilus, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis,
Eikenella corrodens, and Kingella kingii) are mouth gram-negatives that are
coming to be more recognized as infectious agents -- all are hard to grow in the lab
unless you're tipped off this is endocarditis.
(Arch. Path. Lab. Med. 129: 511, 2005), can produce subacute bacterial endocarditis.
The HACEK group (Hemophilus aphrophilus, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis,
Eikenella corrodens, and Kingella kingii) are mouth gram-negatives that are
coming to be more recognized as infectious agents -- all are hard to grow in the lab
unless you're tipped off this is endocarditis.
The distinction between acute and subacute is kind of artificial, of course.
* Culture-negative endocarditis should make you think of coxiella, bartonella, or Whipple's (J. Inf. Dis. 193: 1711, 2006 -- Whipple's is now infamous Am. J. Med. 123: 962e1-4, 2010).
* "Chronic Q-fever" is coxiella endocarditis and the patient will be on antibiotics for maybe three years. Remember that one inhaled organism can cause disease, it's durable in the environment (both facts that make it a good bioterrorism agent), you'll make the call on serology, it may not be clear how the animal contact happened, and that it lives in macrophages.
Clinicians spend a great deal of time looking for endocarditis, which is good. It's notoriously subtle in producing symptoms, it is treatable, and it is fatal if untreated. The mainstay of making the diagnosis is the positive blood culture, coupled with findings on daily meticulous physical exam (changing murmurs, petechiae, etc.)
If some Hippocrates has given your patient antibiotics "for that fever", don't expect to grow the bacteria even if the vegetations are massive.
*Q-fever![]() endocarditis: Am. J. Med. 100: 629, 1997. Diagnosis by serology.
endocarditis: Am. J. Med. 100: 629, 1997. Diagnosis by serology.
{06563} acute bacterial endocarditis
{08458} acute bacterial endocarditis
{38563} bacterial endocarditis
{03293} bacterial endocarditis, aortic valve cusp perforation
{03413} bacterial endocarditis, gross
{06986} bacterial endocarditis
{06989} bacterial endocarditis
|
|
NON-BACTERIAL THROMBOTIC ENDOCARDITIS ("marantic endocarditis")
This is a non-bacterial accumulation of fibrin and platelets on the lines of closure of valves. Individual vegetations are seldom more than 5 mm across. Tissue pathology is reviewed in Mayo Clin. Proc. 76: 1204, 2001.
Classically patients have been cancer victims (notably pancreatic cancer), but more recently the syndrome has been appearing in patients with autoimmune disease, notably lupus anticoagulant.
Sometimes these contain a few polys, but usually they don't. (The fact that they are not inflamed lets you distinguish them from acute rheumatic fever.) Sometimes they embolize (for example, Ann. Thor. Surg. 79: 2145, 2005), but usually they don't.
{06326} Non-bacterial thrombotic endocarditis
{06965} Non-bacterial thrombotic endocarditis
{53315} Non-bacterial thrombotic endocarditis
{53317} Non-bacterial thrombotic endocarditis
|
|
LUPUS ENDOCARDITIS ("Libman-Sacks")
Lupus and/or the antiphospholipid syndrome (Circulation 93: 1579, 1996) can result in small masses of fibrin and a bit of inflammation on various valve surfaces and anywhere else on the endocardium.
It seems reasonable to think that these are foci of type III immune injury. Be this as it may, you might spot a hematoxylin body here.
Lupus patients are living longer and being studied more closely. We now know that Libman-Sacks is anything but benign. Valves often end up mutilated (Am. J. Med. 120: 636, 2007) much like rheumatic fever.
{06962} Libman-Sacks endocarditis
![]() Libman-Sacks Endocarditis
Libman-Sacks Endocarditis
Thailand
CALCIFICATION OF THE MITRAL ANNULUS
A common, minor problem seen in many older patients. While various rhythm disturbances and mitral insufficiency are attributed to this lesion, most often it seems harmless. A few pathologist will attribute sudden death to it if it's obviously roughing-up the AV node.
![]() Calcified mitral valve annulus
Calcified mitral valve annulus
WebPath photo
CARCINOID HEART DISEASE (Prog. Card. Dis. 49: 439, 2007; Am. J. Card. 107: 1221, 2011)
Carcinoids that release their products into the great veins (i.e., those in the ovary or metastatic in the liver) often produce endocardial fibrosis, generally limited to the right side of the heart (i.e., whatever does it is "detoxified" in the lung). The tricuspid and pulmonic valves may malfunction, with tricuspid regurgitation being the usual finding.
The tricuspid and pulmonic valve leaflets are likely to thicken; the pulmonic leaflets stick together and the tricuspid leaflets tend to become stuck to fibrous tissue on the nearby endocardium.
Though much of "carcinoid syndrome" is caused by serotonin and bradykinin, it's still not clear which molecules are responsible. The Fenphen fiasco has focused attention back on serotonin, and the higher the 5-HIAA (serotonin metabolite) secretion in the urine, the worse the heart disease (NEJM 348: 1005, 2003).
{08044} carcinoid heart disease (trust me; look for the
white fibrous stuff)
{08047} carcinoid heart disease (pulmonic valve, trust me)
ENDOMYOCARDIAL FIBROELASTOSIS ("endocardial fibroelastosis", etc.)
Some infants in the U.S. develop endocardial fibroelastosis, a tough thickening of the endocardium
that ultimately interferes with the heart's ability
to contract. There are a variety of uncommon causes, but the disease
has nearly vanished thanks to the elimination of
mumps![]() ,
which crossed the placenta and infected the unborn child (Circulation 95: 133, 1997.)
,
which crossed the placenta and infected the unborn child (Circulation 95: 133, 1997.)
Especially in central Africa, there's a relatively common, similar lesion centered on the apex. The fibrosis stiffens the entire heart, and eventually leads to mitral and tricuspid insufficiency. In the past, this has been blamed on eating little green bananas ("rich in serotonin, mimicking the carcinoid syndrome"); however I understand these are also a staple in the Caribbean and that the disease isn't a well-recognized problem there as it is in tropical Africa. I have been unable to find anything recent about its likely causes.
The truth is that the disease is greatly under-recognized, with perhaps ten million cases worldwide, making it by far the most common "restrictive" heart disease. Survival is around two years. See NEJM 359: 43, 2008.
{10739} endocardial fibroelastosis
* WHIPPLE'S ENDOCARDITIS is a fibrosing process caused by Tropheryma whipplei which is familiar from the gut. Bacteria fill macrophages which in turn fill and deform the endocardial tissue with resulting scarring. See J. Inf. Dis. 190: 935, 2004.
Q-FEVER![]() endocarditis, caused by the rickettsia, is also well-known
(J. Inf. Dis. 187: 1097, 2003.
endocarditis, caused by the rickettsia, is also well-known
(J. Inf. Dis. 187: 1097, 2003.
COMPLICATIONS OF ARTIFICIAL VALVES: common and serious. Nobody makes valves as good as the Mother Nature's, but sometimes the originals do need to be replaced. Ask a cardiac surgeon about these problems.
{53323} Bjork-Shiley "toilet seat"/"disk" valve
{03626} infection on prosthetic valves
{07732} prosthetic valve endocarditis
{07723} worn-out prosthetic valve
MYOCARDITIS (Lancet 379: 738, 2012)
|
|
|
Inflammation of the heart. The heart muscle itself is incredibly resistant to bacterial infections unless they extend from endocarditis. Most myocarditis is probably viral and/or autoimmune.
The only systemic bacterial infection that is likely to produce a neutrophilic myocarditis is meningococcemia, and this seems to be the exception even with this dread micro-organism. The well-known negative inotropic effect in meningococcemia now seems to be mediated by interleukin 6 (Lancet 363: 203, 2004).
Viruses damage heart cells both directly (cytopathic effect) and by
causing T-cells to attack them. Most worthy of note are coxsackie A![]() and coxsackie B
and coxsackie B![]() virus infections (look for single-fiber
necrosis; remember the selenium deficiency in China made people much
more vulnerable to this), parvovirus B19 (has been under-recognized
as it produces no distinctive morphology: NEJM 362: 1248, 2010), diphtheria (remember the toxin?), trichinosis (look for eosinophils, even if you see no
worms), anthracyclines (adriamycin-doxorubicin; classic work Am. J. Clin. Path. 100: 158,
1993; Ann. Int. Med. 125: 40, 1996) and Chagas' disease (look for the T. cruzi bugs in the heart
cells, and/or an "autoimmune" cardiomyopathy kept going by the presence
of a very few protozoans: Am. J.
Card. 79: 1135, 1997 -- tropical medicine buffs know this can also happen in the eastern
African strain of sleeping sickness).
virus infections (look for single-fiber
necrosis; remember the selenium deficiency in China made people much
more vulnerable to this), parvovirus B19 (has been under-recognized
as it produces no distinctive morphology: NEJM 362: 1248, 2010), diphtheria (remember the toxin?), trichinosis (look for eosinophils, even if you see no
worms), anthracyclines (adriamycin-doxorubicin; classic work Am. J. Clin. Path. 100: 158,
1993; Ann. Int. Med. 125: 40, 1996) and Chagas' disease (look for the T. cruzi bugs in the heart
cells, and/or an "autoimmune" cardiomyopathy kept going by the presence
of a very few protozoans: Am. J.
Card. 79: 1135, 1997 -- tropical medicine buffs know this can also happen in the eastern
African strain of sleeping sickness).
Also worth mentioning are the rickettsial diseases:
typhus![]() ,
Rocky Mountain spotted fever
,
Rocky Mountain spotted fever![]() ,
Q-fever
,
Q-fever![]() .
.
For research, you'll want to see necrosis of myocaridal fibers plus a lymphocytic infiltrate to make the call of "active myocarditis" ("the Dallas criteria").
You may find abscesses in the heart muscle in people dying of sepsis, or with severe diabetes.
|
|
{24946} coxsackie B![]() myocarditis
myocarditis
{03461} myocarditis, microscopic
{15753} Chagas' disease
{26582} Chagas' disease
{53320} toxoplasmosis![]()
|
|
|
|
![]() Chagas cardiomyopathy
Chagas cardiomyopathy
CDC
Wikimedia Commons
In most of these forms of myocarditis, look for a lymphocytic infiltrate coupled with various kinds of damage to myocardial fibers.
Loeffler's eosinophilic disease of the heart usually features fibrosis of the endocardium with a lot of eosinophils (often a restrictive cardiomyopathy). It is part of the "hypereosinophilic syndrome", a mixed bag of myeloproliferative diseases, worm infestations, and "idiopathic" -- regardless of cause (if it can be found), Loeffler's now treatable with the anti-IL-5 antibody mepolizumab (NEJM 358: 1215, 2008).
Sarcoidosis produces a cardiomyopathy in which much of the heart muscle is replaced by non-caseating granulomas and fibrosis. It's quite common in the illness and probably gets missed a lot (Chest 133: 1426, 2008). Sarcoidosis identified on heart biopsy is ominous (Am. Heart J. 150: 459, 2005).
Hypersensitivity myocarditis is quite common, usually due to medications. You'll see eosinophils in the heart muscle, especially around the vessels.
"Idiopathic giant-cell myocarditis" (* formerly "Fiedler's myocarditis") is a thankfully-rare disease, mostly of young men. The giant cells are probably of muscle origin and there are no granulomas. It's likely that Fiedler's is a reaction pattern, at least sometimes caused by autoimmunity, very deadly and with variable responses to immunosuppression. See Mayo Clin. Proc. 74: 1221, 1999. In humans, the eye muscles are prone to be involved also. The animal model involves immunizing with myosin.
{07946} sarcoid heart
![]() Idiopathic giant cell myocarditis
Idiopathic giant cell myocarditis
Pittsburgh Pathology Cases
Also remember: As you reject your heart transplant, it will become inflamed.
As you would expect, all lead to a dilated, failing heart in severe cases.
CARDIOMYOPATHIES: Non-inflammatory, non-ischemic heart muscle disease. (Call the heart transplant team!) Review (still good): Br. Med. J. 315: 1529, 1997.
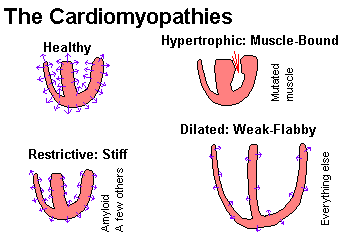
ARRHYTHMOGENIC RIGHT VENTRICULAR CARDIOMYOPATHY ("arrhythmogenic right ventricular dysplasia"; Uhl's; "parchment right ventricle"; J. For. Sci. 42: 32, 1997; Circulation 94: 983, 1996; Arch. Path. Lab. Med. 129: 1330, 2005; South. Med. J. 101: 309, 2008; Am. Fam. Phys. 73: 1391, 2006; Lancet 373: 1289, 2009; Lancet 385: 662, 2015. sudden death AMFJP 30: 78, 2009 progressive nature of the lesion with apoptosis Am. J. Path. 152: 479, 1998; sudden death in vigorous good health AJFMP 18: 345, 1997).
The pathologist sees a curious mix of fatty ingrowth and fibrosis in the wall of the right ventricle (sometimes the left also or instead). We're working out the pathology and mechanism. The cell pathology is bizarre (Am. J. Med. Sci. 320: 310, 2000). First series of 200 deaths: Circulation 108: 3000, 2003.
Today's diagnostic imaging specialist must be familiar with this entity. You'll follow-up suspicious EKG"s with MRI's, which show both the fat (less important, since it's often mostly fibrosis instead of fat) and the dysfunction (more important). Update Am. Heart. J. 155: 147, 2008.
|
|
|
|
|
|
|
|
|
|
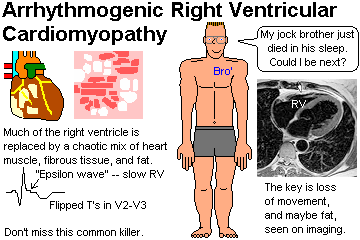
DILATED ("flabby heart"; "congestive cardiomyopathy")
{03629} dilated cardiomyopathy vs. normal
{03632} dilated cardiomyopathy
{07769} dilated cardiomyopathy
{11555} dilated cardiomyopathy
![]() Dilated Cardiomyopathy
Dilated Cardiomyopathy
CDC
Wikimedia Commons
There are a host of lesions that cause the heart muscle to lose its strength. There's a list in "Big Robbins"; most cases never get a diagnosis (NEJM 331: 1564, 1994; still probably true). Only 50% of cases of dilated cardiomyopathy even have a diagnosable lesion on microscopy.
In the dilated cardiomyopathies, the heart usually weighs more than "normal", but it is very distended and thus the wall is usually of normal thickness or even unusually thin. (Why?)
ALCOHOLIC'S HEART
Alcohol is rough on most organs, and the liver and brain damage take precedence most of the time to the cardiac toxicity.
Alcohol may or may not have a direct toxic effect on the heart, by itself or through a metabolite.
Lots of alcoholics are deficient in vitamin B1, and this can produce high-output failure or even weaken the muscle.
In 1966, some folks added cobalt to Quebec beer to make a nicer head of foam. Cobalt poisons pyruvate metabolism and produced a beriberi-like cardiomyopathy. (* The classic pathology: Ann. N.Y.Acad. Sci. 156: 577, 1969).
Selenium deficiency led to an epidemic of dilated cardiomyopathy in China, and occurs in patients in the developed world who suffer from malabsorption (Med. Sci. Law 42: 10, 2002).
PREGNANCY
Around the time of parturition and/or up to six months after, some women get a dilated cardiomyopathy. This "peripartum cardiomyopathy" is very serious: JAMA 283: 1183, 2000 (NIH conference).
Sadly, this is mostly a disease of poor women, especially those who have had several babies close to one another. Perhaps it is the result of a deficiency in some trace nutrient. It remains mysterious: Am. Heart J. 130: 860, 1995; and it's likely to recur: NEJM 344: 1567, 2001.
* Today, some people believe prolactin inhibition should be used as part of the treatment (Ann. Thor. Surg. 91: 274, 2011). There are a variety of empirical treatments, the disease can hang around for a long time, and several percent of women still die of it
ACROMEGALY (apoptosis of myocytes for some reason): Circulation 99: 426, 1999.
POLYMYOSITIS-DERMATOMYOSITIS can produce a cardiomyopathy, accelerate atherosclerosis, and/or promote widespread small-artery disease, depending on the patient (Rheumatology 45 S4: iv18, 2006.)
|
"POST-VIRAL", allegedly autoimmune ("Barney Clark's disease").
Lots of people with "end-stage idiopathic dilated cardiomyopathy with lymphocytes" have some lab
evidence of autoimmunity against the heart (specifics vary, and will bewilder you), and "a possible
history of viral myocarditis". Actually, the link between this entity and viral infection is pretty
weak. Many of these people have antibodies against coxsackie To nobody's surprise, the lymphocytes turn out to be T-cells, and unlike those in a normal person's heart, most of these seem to be active (Circulation 92: 2876, 1996). * Update on autoimmunity and viruses, and how they may work together, in causing "idiopathic cardiomyopathy": Nat. Med. 9: 1448, 2003. Autoantibodies against the beta-1 adrenergic receptor are also fairly common in idiopathic dilated cardiomyopathy. These seem to induce apoptosis (Circulation 116: 399, 2007). Here's a classification scheme for "idiopathic, probably viral myocarditis":
BORDERLINE MYOCARDITIS: Enough lymphocytes, but you can't really find those necrotic fibers FULMINANT MYOCARDITIS: The left ventricle fails, but is NOT dilated yet. On biopsy, there are lots of white cells, mostly neutrophils. The patient will either recover or die fast. CHRONIC ACTIVE MYOCARDITIS: Just as in the liver, there are patches of lymphocytes and ongoing fibrosis, over years, progressing to end-staged dilated cardiomyopathy EOSINOPHILIC MYOCARDITIS: The "acute" form mentioned above (due to pre-transplant meds), drug allergy, or one of the eosinophil-rich diseases like Loeffler's, or CMV in the immunocompromised, or toxoplasmosis or Chagas's or who-knows-what? GIANT-CELL MYOCARDITIS: Fiedler's. The giant cells are of myocyte origin, and there are no granulomas. The prognosis is ominous without treatment. |
 Barney Clark |
GENETIC SYNDROMES: Syndromes worth remembering for general medicine include Friedreich's ataxia and Duchenne's-Becker's muscular dystrophy. Even female carriers of the latter often get a dilated cardiomyopathy (JAMA 275: 1335, 1996).
Lately we've noticed that around 15% of idiopathic dilated cardiomyopathy patients have a mutation in one of the genes that produce desmosomes (Heart 97: 1744, 2011). Stay tuned.
There are some other syndromes that give only a dilated cardiomyopathy (* one is due to mutant actin: Circulation 99: 1022, 1999; others to mutant troponin T (pathology Circulation 104: 1380, 2001), and beta-myosin heavy chain NEJM 343: 1688, 2000; mutated desmin affects skeletal and cardiac muscle NEJM 342: 770, 2000; phospholamban Science 299: 1410, 2003; troponin I: Lancet 363: 371, 2004 is unusual for being recessive). Nexilin, a Z-band protein: Nat. Med. 15: 1281, 2009.
"Noncompaction cardiomyopathy" ("spongiform cardiomyopathy"; Circulation 108: 2672, 2003) features very much enlarged trabeculae with blood lakes in the wall or one or both ventricles. Easy to spot on echo (Am. J. Card. 94: 389, 2004), it is is acquired during embryogenesis, either with a known mutation / syndrome or by itself. It can produce a dilated or a restrictive pattern. Mutation at NOTCH: Nat. Med. 19: 193, 2013.
* "Histiocytoid cardiomyopathy" is a rare pediatric disorder with striking infiltration by abnormal myocardial cells that look like macrophages; they have abnormal mitochondria. The loci are being sought. This is one of the reasons it's a good idea to look at the histology of the heart in a "SIDS" case.
Remember sarcoidosis, Chagas, radiation, giant-cell myocarditis, rheumatic fever, ipecac (Munchausen's by proxy: Pediatrics 97: 902, 1995).
"Stress cardiomyopathy / myocardial stunning / transient apical ballooning syndrome / broken heart syndrome", after a jolt of epinephrine, has been precipitated even by surprise parties (NEJM 352: 539, 2005) or opiate withdrawal (Mayo Clin. Proc. 81: 825, 2006). The quaint term for this illness, which features ballooning of the ventricle especially at the apex, raised cardiac enzymes, and generally a good outcome, is "takotsubo", Japanese for "octopus trap". There's one report of rupture after a quarrel (literally dying of a broken heart: Mayo Clin Proc. 79: 821, 2004). Some folks do die of it. Update JAMA 306: 277, 2011; NEJM 373: 929, 2015.
Seldom fatal, but worth knowing: In the most severely injured patients, notably those with SEVERE BURNS, there is weakening of the heart; in the animal model, there is widespread apoptosis of cardiac myocytes (J. Trauma 65: 401, 2008).
Seldom fatal, but worth knowing: A common cardiac lesion in patients with chronic renal failure (UREMIC CARDIOMYOPATHY) is fibrosis around individual fibers throughout large areas. This greatly enlarges the left ventricle, but doesn't really interfere so much with function at least during systole. Update Kid. Int. 69: 1839, 2006. People who've been on hemodialysis for a long time often have calcium chunks in the heart; these can be huge but seldom cause problems (Medicine 91: 165, 2012).
* The old story about the earlobe crease as a marker for precocious atherosclerosis holds up on the Swedish autopsy service: AJFMP 27: 129, 2006.
* Still controversial is whether diabetes itself causes a cardiomyopathy, perhaps by increasing the connective tissue among the myocardial cells. See Heart 92: 296, 2006; J. Am. Coll. Card. 47: 693, 2006).
* The left-ventricle assist device (plus a medical regimen) sustains patients with cardiomyopathy as it heals over time. This now seems to be a life-saver, and will probably be fairly common when you are in residency. See NEJM 355: 1873 & 1922, 2006.
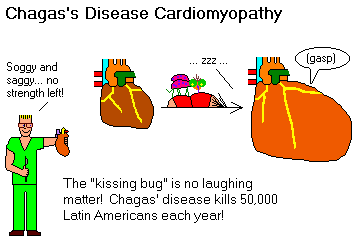
HYPERTROPHIC CARDIOMYOPATHY(Lancet 381: 242, 2013; "idiopathic septal hypertrophy", "asymmetric septal hypertrophy"; "Les Aspin's disease"; "Reggie Lewis's disease"; "muscle-bound heart"; Hank Gathers supposedly had myocarditis rather than hypertrophic cardiomyopathy NEJM 329: 55, 1993)
|
{07811} hypertrophic cardiomyopathy
|
This curious lesion, generally autosomal-dominant (13 loci, all of them cardiac sarcomere proteins), exhibits (1) variable regions of hypertrophy (in contrast to the "concentric hypertrophy" of left ventricular overwork and the "eccentric hypertrophy" of left ventricular overfilling); (2) disarray of cardiac muscle cells by light and electron microscopy (often perpendicular / oblique to one another); (3) fibrosis around muscle cells; (4) some fibrous narrowings of the small arteries.
This is serious. The outflow tract from the left ventricle toward the aortic valve is likely to be obstructed by the large mass of muscle ("obstructive hypertrophic cardiomyopathy"; "HOCM") and the way it pushes on the anterior leaflet of the mitral valve.
Be careful! If you do aerobic exercise and develop your heart muscle, you're likely to make this particular problem worse. (Here's the case for routine auscultation of the heart in would-be athletes. See J. Fam. Pract. 52: 127, 2003 for the pre-sports physical, author recommends what I've always done and taught.) Same if you get fat (Am. J. Card. 112: 1182, 2013). The septum typically takes its thick shape in the teenaged years. Like other causes of anatomic or functional aortic stenosis, patients are troubled with exercise intolerance, angina, syncope, and/or sudden death. Infamously, sudden death can be the first "warning".
You'll learn about the murmur, which sounds like aortic stenosis but is usually at the LLSB and seldom radiates to the neck, on rotations. Increasing chamber volume makes the murmur softer, in contrast to other murmurs (why?)
Most cases are autosomal-dominant, and a majority are caused by a mutated beta-myosin gene. Mouse with the mutation: Science 271: 663, 1996. Other loci are now known, mutant troponin T is common and especially deadly (histopathology Circulation 104: 1380, 2001), and tropomyosin and myosin binding protein are rare (NEJM 332: 1058, 1995; J. Am. Coll. Card. 29: 635, 1997; NEJM 338: 1249, 1998; Lancet 355: 58, 2000).
* Danon's cardiomyopathy (gene LAMP2; JAMA 301: 1253, 2009 -- one of those rare X-linked semi-dominant diseases), a systemic disease with glycogen storage vacuoles, can produce an extreme hypertrophic cardiomyopathy, with or without an extra-thick septum / crisscross fibers.
Many patients die suddenly in youth. But many others do not die of the disease, or die of it in old age (CHF or rhythm problems: JAMA 281: 650, 1999).
* When in doubt about how to manage this, put the patient on a treadmill. Dropping the blood pressure is a warning of more ominous things to come. The risk for sudden death cannot be predicted simply by measuring the thickness of the wall (Lancet 357: 420, 2001). Gadolinium scan to predict risk: J. Am. Coll. Card. 41: 1561, 2003.
The implantable defibrillator for these people: NEJM 342: 422, 2000.
Today, alcohol ablation of the obstructing muscle mass (i.e., alcohol injected into the coronary arteries) is a well-established therapy for patients not able to tolerate the surgery. Pathologists see Am. J. Card. 99: 563, 2007 and especially Heart 92: 1773, 2006 (coagulation necrosis of the coronary arteries themselves). As you'd expect, heart block may be unavoidable.
* Forme frustes? There are several genes (sarcomeres, mitochondria) which produce rare genetic cardiomyopathies. In the Framingham study, about person in 30 has unexplained, mild left ventricular hypertrophy and of these, one in 5 is heterozygous for one of these (Circ. 113: 2697, 2006); we await full data on the prevalence of these mutations in those without hypertrophy, but this is worth watching.
* The name of Reggie Lewis will always be associated with hypertrophic cardiomyopathy. However, this was not found on ultrasonography in life, or at autopsy. The extensive scarring described at autopsy sounds much more like "cocaine heart". During the litigation, which finally ended in 2005, there was a great deal of lawyering by attorneys for his family about Lewis's failing and/or refusing to take drug tests for cocaine. Of course, we'll never know for certain.

RESTRICTIVE-INFILTRATIVE-OBLITERATIVE ("stiff heart"; usually amyloid)
Most older people get some amyloids of various compositions (known and unknown) in their atria and aortas. All about amyloid and the heart: J. Clin. Path. 58: 125, 2005; clinicians see Am. J. Med. 124: 1006, 2011.
If amyloid involves the myocardium extensively, the muscles cannot contract. This is the usual cause of "restrictive cardiomyopathy" in the developed world. Cardiac amyloidois Am. J. Med. 124: 1006, 2012.
* Amyloid hearts bounce; I was shown this once. Don't try it; it's disrespectful.
* Fabry's storage disease produces a restrictive cardiomyopathy, and sometimes a very severe case of sarcoidosis or hemochromatosis can do the same.
* A small minority of hypertrophic cardiomyopathy patients (with the same genes as the classic presentation) have a stiff heart with little hypertrophy (J. Am.Coll. Card. 49: 2419, 2007); the prognosis is of course worse than in the classic syndrome.
Some "restrictive cardiomyopathy" patients, both children and adults, have developed an idiopathic interstitial fibrosis (i.e., collagen rather than amyloid) that makes the heart stiffer.
When super-iron-overloaded, the heart of a hemochromatosis patient can present as restrictive disease.
We've already seen endocardial fibroelastosis as a mimic, and some of the diseases of skeletal muscle can also produce a restrictive cardiomyopathy (Heart & Lung 40: e123, 2011).
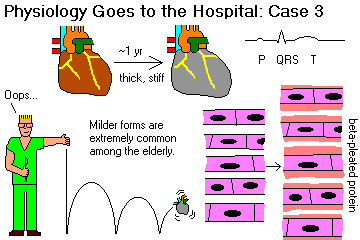
{07937} amyloid heart
{07940} amyloid heart
{07943} amyloid heart
SECONDARY CARDIOMYOPATHIES: Hemochromatosis (dysrhythmogenic!), hypothyroidism (low-output, myxedema / extra ground substance in the wall of the heart, and without thyroid hormone the heart muscle cells don't want to beat), pheochromocytoma ("catecholamine heart", with single-fiber necrosis/apoptosis as in cocaine heart and perhaps after burns), chemotherapy (doxo- and daunorubicin; "Adriamycin"; flabby dilated heart), Pompe's glycogenosis, other glycogen storage diseases (glycogen synthetase deficiency NEJM 357: 1507, 2007), Duchenne's (dilated cardiomyopathy), Friedreich's (a variety of phenotypes Circulation 125: 1626, 2012; as a mimic for hypertrophic cardiomyopathy -- Irish J. Med. Sci. 180: 799, 2011), end-stage HIV infection (Br. Med. J. 309: 1605, 1994).
Again, Fabry's is worth knowing, because it can mimic hypertrophic or restrictive cardiomyopathy, and is treatable with enzyme replacement (Circulation 105: 1407, 2002). Around 0.5% of "hypertrophic cardiomyopathy" patients turn out to have Fabry's (Heart 97: 1957, 2011).
Adriamycin is hated for producing a characteristic dilated cardiomyopathy. You may see pale-blue-staining myocardial cells without sarcomeres ("adria cells") next to normal cells. Adriamycin damages the mitochondria and somehow complexes with iron, producing free radical injury to the surrounding sarcomeres (J. Clin. Inv. 117: 3730, 2007). Anyone treated with adriamycin is likely to have fibrosis between the myocardial cells for the rest of their lives -- future pathologists be aware of this.
I'm not big on the term ISCHEMIC CARDIOMYOPATHY for the fibrosis of the heart that's sustained many little infarcts, since the disease is not primarily the muscle. It's come into general use, though, and it's diagnosable on imaging -- remember that it remodels (Am. J. Card. 109: 390, 2012).
{07039} von Gierke's disease
{07958} hemochromatosis
{07967} Fabry's
{08053} pheochromocytoma heart
{08078} radiation atherosclerosis
{08081} radiation pericarditis
* CADAVERIC SPASM
This is instantaneous rigor mortis, generally seen in extreme pain or fright. It's said that the heart can stop as well in this setting. The mechanism must be influx of calcium into skeletal and cardiac muscle, as in rigor mortis.
There's no sorting this out, or saying whether this is "on a continuum with stunned myocardium."
This is especially helpful in drownings, where water weeds held tight in the hands are proof that the person was alive in the water.
There are old reports from the battlefield of soldiers found dead, or seen to die, with no injury, but with their muscles frozen in position, generally after a shell had exploded nearby. The cause was supposedly terror. Since the accounts are of groups of soldiers all dying at once, your lecturer believes these were blast injuries, with internal damage not visible on the outside, and that the bodies were found after rigor had developed normally.
If cadaveric spasm really occurs, it must be rare. It makes for good stories, though. It's also the origin of the comforting untruth that, if you fall or jump off a building or something and see that you will surely die, your heart will stop before you hit the ground.
PERICARDIAL DISEASE (Lancet 363: 717, 2004)
|
|
PERICARDIAL EFFUSIONS
Many different situations result in accumulation of fluid (i.e., more than the usually few mL) in the
pericardial sac. Viral pericarditis is often accompanied by a serous effusion; TB's![]() effusion may also
be serous.
effusion may also
be serous.
However, to produce tamponade, it takes around 150 mL of fluid, and it must build up pressure rapidly (i.e., hemopericardium after ventricular rupture, penetrating trauma, or aortic dissection; pus in a suppurative pericarditis). If the fluid accumulates slowly, the pericardium can expand. I've seen a one-quart effusion that didn't seem to cause a problem.
* "Recurrent pericarditis" is a nuisance illness that seems to do little harm; your lecturer had long believed that it is a variant of familial mediterranean fever and this has been borne out now by the striking success of colchicine to end attacks (Am. Fam. Phys. 85: 1098, 2012).
On rotations, you'll learn how to do pericardiocentesis, and to place a pericardial window to keep the fluid "off the heart".
PERICARDITIS
Inflammation of the pericardium hurts. Patients typically get relief by leaning forward. You, the physician, will strain to hear the notoriously ephemeral "friction rubs" of this illness.
In sporadic pericarditis, the etiologic agent is probably some virus or immune phenomenon. It usually is self-limited; today people treat it with NSAIDs. Colchicine is the second drug; try both before moving to a glucocorticoid (JAMA 314: 1498, 2015).
"Fibrinous pericarditis" (the familiar "bread and butter", or more realistically, "ketchup on bread" lesion) results from myocardial infarction (over a transmural infarct; Dressler's), uremia, radiation, lupus, Behcet's (surprisingly common Medicine 91: 25, 2012), rheumatic fever, surgery, and trauma. It may organize (obliterating the pericardial space, which isn't usually a big deal), or even calcify (perhaps limiting distensibility).
{18719} fibrinous pericarditis
|
|
|
Pus in the pericardial space usually means bacterial infection. They usually got there by way of a
nearby pneumonia or empyema, though there are other routes. Again, it may organize (a few
adhesions, or "obliterative pericarditis"; or even calcify. TB![]() in particular is infamous for causing
troublesome scars. Old healed pericarditis (or TB, or asbestos, or amyloid
selectively involving the pericardium, or "idiopathic")
may become "constrictive" and cause problems.
in particular is infamous for causing
troublesome scars. Old healed pericarditis (or TB, or asbestos, or amyloid
selectively involving the pericardium, or "idiopathic")
may become "constrictive" and cause problems.
![]() Suppurative pericarditis
Suppurative pericarditis
WebPath photo
"Hemorrhagic pericarditis" is due almost exclusively to TB![]() and cancer.
Caseous
and cancer.
Caseous![]() material in the
pericardial sac is probably TB.
material in the
pericardial sac is probably TB.
{03659} constrictive non-calcific pericarditis
* The "soldier's plaque" or "milk spot" on the visceral pericardium (i.e., the epicardial surface) is a focus of very slight thickening of the fibrous layer under the mesothelium. This mysterious finding is harmless. It was once attributed to packs worn during WWI, until some clever person noted that it's equally common in women. Today, somebody may tell you it represents "healed pericarditis", but it doesn't really look like scar.
CARDIAC TUMORS are rare.
The principal primary heart tumor is the MYXOMA, a lesion of endothelial origin that arises as a ball from the atrial septum and fills the left atrium. Grossly, it's a typically benign, soft tumor. Microscopically, there are lots of spindle cells and ground substance, and few vessels. Myxomas (familiarly called "wrecking balls") can plug the mitral valve (instant death), or cause emboli (under-recognized, Chest 107: 674, 1995).
Atrial myxoma are really tumors, not just organized atrial "ball-valve" thrombi of the sort seen in mitral valvular disease. They're clonal, and have a few antigenic markers.
* Being genuine tumors, there's an important anti-oncogene deletion syndrome ("Carney complex", with lentigos and gland overfunctions too, and others: Am. J. Card. 79: 994, 1997). If you see a myxoma in the right atrium rather than the left, suspect a familial syndrome.
![]() Atrial myxoma
Atrial myxoma
AFIP
Wikimedia Commons
People with tuberous sclerosis are prone (~50%) to have multiple small cardiac hamartomas ("rhabdomyomas"). These are occasionally troublesome. Even in kids without TS, these are the most common heart tumor; they are prone to regress over time.
* The other pediatric tumor of the heart is the troublesome cardiac fibroma, which juts off the septum and interferes with outflow. Thankfully, it's rare.
I was amazed by my first PAPILLARY FIBROELASTOMA (J. Thor. Card. Surg. 112: 551, 1996; fatal AJFMP 19: 162, 1998) at a 1991 autopsy. It looked like a kooshball on the pulmonic valve. These form on any valve leaflet. Depending on location, they can cause embolic stoke (Clin. Card. 24: 346, 2001); look for them using transesophageal echocardiography. Series Circulation 103: 2687, 2001; surgery Ann. Thor. Surg. 80: 1712, 2005. Smaller, similar-looking lesions on the aortic leaflets called LAMBL'S EXCRESCENCES are supposed to be organized thrombi; they occasionally embolize too.
Lipomas ("lipomatous hypertrophy" / "massive fatty deposits") of the septum are occasionally seen (review AJFMP 17: 43, 1996); if it interferes with flow, it can be removed.
* Intracardial leiomyomatosis is the presence in a cardiac chamber of a leiomyoma that has extended directly from a vein -- it is common enough to have a surgical literature (J. Thor. Card. Surg. 142: 823, 2011).
* "Cystic tumor of the AV node" is a hamartoma that mimics the ultimobranchial nests of the thyroid (Am. J. Clin. Path. 123: 369, 2005).
{08029} lipomatous hypertrophy ("lipoma") of atrial septum
* The AV node is subject to a poorly-understood, rare lesion called "cystic tumor" (formerly "mesothelioma", a misnomer). Epithelially-lined cysts are filled with mucin. Heart block can result.
Primary cancers of the heart (i.e., sarcomas) are so rare that even Mayo's sees only about one a year; of course, they are deadly (Cancer 112: 2440, 2008.)
Metastases to the heart are usually from lung, breast, melanoma, lymphoma, leukemia, and choriocarcinoma. Any of these can be troublesome (heart block, muscle failure, effusion), but you will usually miss the diagnosis during life.
LAST PHOTOS
{17416} tiger-stripes of fatty change in extreme anemia; remember this one?
{11480} thromboembolus caught in heart

PULSUS PARADOXUS OVERSIMPLIFIED
You need to understand this. In health, venous pressure drops slightly during inspiration because the decreased intrathoracic pressure draws venous blood into the chest.
In health, arterial pressure drops slightly during inspiration because the greatly expanded pulmonary vascular bed takes up the right cardiac output, diminishing return to and output from the left. The resulting systemic arterial pressure drop is less than 10 torr and is not obvious on palpation.
In restrictive pericardial disease, the diaphragm pulling down on the pericardium pulls it even tighter. This further blocks venous return to the heart, so that venous pressure actually increases during inspiration. The neck veins are more distended during inspiration, and drop during expiration. In restrictive pericardial disease, breathing in also blocks arterial outflow from the heart, greatly accentuating the normal drop in arterial pressure during inspiration (>10 torr, should be detectable on palpation).
|
FINISHING UP
* The desires of the heart are as crooked as corkscrews.
--W.H. Auden
By now, one would have to be very naive to think
that most of his or her neighbors (patients, whatever) don't prefer death
to severe and expensive disability. This preference is showcased in a study (NEJM 330: 545, 1994)
in which a group of older patients were told the (abysmal) odds of surviving CPR reasonably intact.
The vast majority never-ever wanted it done to them. CPR doesn't work for
trauma cases (why not?), and even in ischemic heart disease,
the few cases where it does work are very likely to leave the "survivor"
badly brain-damaged.
Really, one's only hope is rapid defibrillation, which thankfully is now
being emphasized (NEJM 348: 2626, 2003).
When the geriatricians trashed "ER" for
showing an excessively-high rate of good outcomes of CPR, the producers
shot back that it's the doctors' fault that patients hear about CPR from, and only from, Hollywood
(no kidding, NEJM 334: 1578, 1994). As a physician, I welcome signs that the "keep them all alive
as long as you can, you make more money" era is finally coming to an end. As a physician, I'm
sorry that most of the impetus for change hasn't come from my fellow-physicians.
*Transgenic pigs are now being bred as heart donors: Br. Med. J. 312: 657, 1996. Since then, there has been a great deal of work, but with publications only in the super-specialty literature. Stay tuned.
Cats look down on us. Dogs look up to us. Pigs treat us as equals.
-- Winston Churchill

You'll hear in ACLS about ELECTROMECHANICAL DISSOCIATION, i.e., the EKG is fine and the heart isn't pumping. In other words, dead with a normal EKG. What would cause this? HINT: Pulmonary emboli, tamponade, no venous return to the heart because of bleeding or widespread vasodilatation, ruptured septum, no calcium. Understand the "why" of each? Can you think of others? Since the only one that you're going to be able to treat during CPR is "no calcium", you'll give calcium by vein. Don't expect....
In our sleep, pain which cannot forget falls drop by drop upon the heart until, in our own despair, against our will, comes wisdom through the awful grace of God.
-- Aeschylus, "Agamemnon"
tr. by Robert F. Kennedy in his extremporaneous eulogy for Martin Luther King
* A final, personal note: The heart is a source for metaphor. My favorite quotations concerning the heart include Jeremiah 17: 9-10 and 31:33 and Koran 91:4. See also the Katha Upanishad for a classic description of the metaphysical heart. And of all the experiences in clinical medicine, the one that this pathologist loves the most is the sound of the beating heart.
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about pathology of the heart to consult these standard textbooks.
In my notes, the most helpful current journal references are embedded in the text. Students using these during lecture strongly prefer this. And because the site is constantly being updated, numbered endnotes would be unmanageable. What's available online, and for whom, is always changing. Most public libraries will be happy to help you get an article that you need. Good luck on your own searches, and again, if there is any way in which I can help you, please contact me at scalpel_blade@yahoo.com. No texting or chat messages, please. Ordinary e-mails are welcome. Health and friendship!

|
|

| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
EXAMINING THE LIVING CARDIOVASCULAR SYSTEM
Correlations with Pathology
Ed Friedlander MD "the pathology guy"
A group of you asked, in lieu of continuing into "lung", to integrate pathology
and the changes you have learned about on physical exam. Pathologists are the
big integrators and "why"-folks in most medical schools, so here goes....
0. Turn off the TV 1. Inspect 2. Palpate 3. Percuss 4. Auscultate
Valsalva: Reduces, then increases, preload
Leg lift: Increases preload
Handgrip: Increases afterload
Inspection! Apex impulse. Where? How big? Any others? Dextrocardia / situs
inversus. Cyanosis? Jugular venous distention? (For a great review on the physical examination
of the jugular veins, see Am. Heart. J. 136: 6 & 10, 1998.
More than 3 cm vertically above sternal angle, high CVP.) Anything else?
Jugular venous pulse peaks demystified (see also South. Med. J. 100: 1022, 2007....

Big "A"'s: think of high pulmonary vascular resistance ("pulmonary
hypertension", why?
Big "V"'s: Tricuspid regurgitation (why)?
Estimating CVP is very helpful. Be sure
you can recognize Kussmaul's sign in the neck veins in tamponade (and explain
how it happens -- nice case Lancet 371: 1810, 2008)
Palpation! Right ("sternal") lift or heave. Big apex beat is "left lift" or
"left heave". Thrill -- palpable murmur. Feel PMI and carotids
simultaneously. Position of PMI can be measured; duration has most to do with
actual hypertrophy.
Percussion!: Bring your sharpie marker. Don't expect this to tell you as much
or as well as a chest x-ray.
Auscultation!: Bell (S3 and S4; mitral stenosis) vs. diaphragm (everything else). The Littman
diaphragm, lightly applied, supposedly hears the low pitches as well as the bell.
You decide. "I can't tell
you, professor, I am listening to diastole!" Inching along. Sitback and put
your elbows on your knees. Roll over on your left side and lean back against
me.
S1. What is it? Can anybody hear T1?
S2. What is it? P2 is delayed a bit during inspiration. Learning to tell if
a sound is split. Exaggerated split (i.e., P2 is always a little too late):
increased pulmonary vascular resistance (why?), left-to-right shunt (why?),
right bundle branch block (why?) Paradoxical split (i.e., A2 is always a
little too late and/or P2 a bit early); left bundle branch block (why?), some
normal folks. Loud A2: systemic hypertension. Loud P2: Pulmonary
hypertension.
S3: Waves-in-the-wall, the heart as kettledrum? "Tennessee". When do
you hear it? Why?
S4: Atrial sound. "Kentucky". When do you hear it? Why?
Ejection click: Little wiggles in the aortic and/or pulmonary valve leaflets as
they open.
Opening snap: Litle wiggles in the stiff leaflets of a tight mitral valve.
Midsystolic click: Barlow posterior leaflet being pulled tense, less when you
increase preload.
Murmurs mean turbulence. Volume of sound is proportionate to force and volume
of turbulent flow.
Systolic murmurs:
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |


 Blood Vessels and Heart
Blood Vessels and Heart
 Enzyme diagnosis of acute MI
Enzyme diagnosis of acute MI
