Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
verify here. |
Cyberfriends: The help you're looking for is probably here.
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- Lung
Lectures follow Textbook
QUIZBANK: Respiratory
Breakdown of the "Respiratory" quizbank items:
Introduction to Lung Pathology 1-42, 170-198
Interstitial Disease 235-249
Lung Cancer 199-234
Tobacco 43-65
Occupational Disease 150-169
Obstructive Disease 132-149
Infectious Disease 66-131

CHEST WALL PROBLEMS
structural... THE CHEST DEFORMITIES
neuromuscular...THE PARALYSIS & WEAKNESS SYNDROMES
OBSTRUCTED UPPER AIRWAY
structural... QUINSY ("PERITONSILLAR ABSCESS"); CROUP ("LTB"")
functional...THE SLEEP APNEAS
OBSTRUCTED LARGE BRONCHI
all, subtotal... CHRONIC BRONCHITIS
one, total... OBSTRUCTIVE ATELECTASIS; ENDOGENOUS LIPID PNEUMONIA
CONSTRICTED SMALL BRONCHI
mast-cell / inflammation mediated... THE ASTHMAS
platelet-mediated... PULMONARY EMBOLUS
apudoma products... CARCINOID SYNDROME
dense collagen... SOME CHRONIC BRONCHITIS VARIANTS
FIBROTIC REPSIRATORY BRONCHIOLES... SILICOSIS; OBSTRUCTIVE BRONCHIOLITIS
COLLAPSED RESPIRATORY BRONCHIOLES... EMPHYSEMA/"CHRONIC BRONCHITIS"
FLUID-FILLED ALVEOLAR SPACES
transudate... ALVEOLAR PULMONARY EDEMA
exudate & pus... THE PNEUMONIAS
exudate, fibrin, debris... THE RESPIRATORY DISTRESS SYNDROMES
surfactant... ALVEOLAR LIPOPROTEINOSIS; ENDOGENOUS LIPID PNEUMONIAS
other lipid... EXOGENOUS LIPID PNEUMONIAS
blood... GOODPASTURE'S DISEASE; ANTI-NEUTROPHIL CYTOPLASMIC ANTIBODY DISEASES, ACUTE MOUNTAIN SICKNESS, OTHER PULMONARY BLEED SYNDROMES
organisms alone... PNEUMOCYSTOSIS, CRYPTOCOCCOSIS
FLUID-FILLED ALVEOLAR SEPTA
transudate... INTERSTITIAL PULMONARY EDEMA
exudate...THE PNEUMONITIS FAMILY; VIRUSES; MYCOPLASMA
FIBROSIS AROUND ULCERATED BRONCHI...BRONCHIECTASIS
FIBROSIS OF ALVEOLAR SEPTA
slow... THE INTERSTITIAL RESTRICTIVE LUNG DISEASES; (idiopathic pulmonary fibrosis, rheumatoid lung, sarcoid, asbestosis, chronic organic pneumoconiosis / hypersensitivity pneumonitis, many others)
fast... THE RESPIRATORY DISTRESS SYNDROMES
COLLAPSED ALVEOLI
large-airway disease... OBSTRUCTIVE ATELECTASIS
alveolar disease... THE RESPIRATORY DISTRESS SYNDROMES
ischemia... PULMONARY EMBOLUS, SEVERE SHOCK plugged airway... ABSORPTIVE ATELECTASIS / ENDOGENOUS LIPID ("golden" / "postobstructive") PNEUMONIA
NECROTIC LUNG ("cavities", etc.)
infarction... PULMONARY EMBOLUS (COMPLICATED)
suppurative... NECROTIZING PNEUMONIAS; LUNG ABSCESS
caseous...![]() TUBERCULOSIS, HISTOPLASMOSIS, BLASTOMYCOSIS, COCCIDIOIDOMYCOSIS
TUBERCULOSIS, HISTOPLASMOSIS, BLASTOMYCOSIS, COCCIDIOIDOMYCOSIS
weird immune... WEGENER'S GRANULOMATOSIS
malignant... LUNG CANCER
PULMONARY ARTERIAL HYPERTENSION
secondary to low alveolar oxygen... see above; also MOUNTAIN DWELLERS
secondary to alveolar fibrosis... see above
Left-to-right shunts / Eisenmenger's
primary... PULMONARY EMBOLUS; VASCULITIS; IDIOPATHIC
HIGH PaCO2... all whole-lung ventilation problems
LOW PO2...
all whole-lung ventilation problems
perfusing non-ventilated lung / blood avoiding the alveoli altogether
fluid/fibrosis in alveolar septa
 MOUNTAIN DWELLERS
MOUNTAIN DWELLERS
STUDY OBJECTIVES
Describe the essential gross and microscopic anatomy of the airways, from trachea to alveolar sacs. Distinguish the two principal types of pneumocytes (I and II). Describe the anatomic and functional barrier to gas exchange at the alveolar-capillary level.
Describe the factors that influence PaCO2 and PaO2. Describe how PaO2 correlates with the actual oxygen content of the blood. Give the conditions when cyanosis will appear.
List the principal causes of edema in the lung, and compare these to things that cause edema anywhere else in the body. Distinguish interstitial and alveolar edema. Explain why edema of the lung is bad for one's health.
Describe pulmonary congestion, and mention its pathologic sequelae.
Review the pathology and pathophysiology of pulmonary thromboemboli. Describe their frequency and clinical correlations.
List the common causes of increased resistance in the pulmonary arteries (the usual cause of "pulmonary hypertension"), and other causes of pulmonary hypertension. Explain why these things are so harmful. Explain hypoxic pulmonary vasoconstriction, why it is useful in health, and why it is such a problem in disease.
Define acute / adult respiratory distress syndrome and list a few of the many synonyms. Tell about the etiologies, gross and microscopic pathology, the pathophysiology, and the common clinical picture.
Explain the pathophysiology and clinical correlations of neonatal respiratory distress syndrome.
Define atelectasis. Tell how lung collapses due to obstruction, compression, airway obstruction, and lack of surfactant, and give clinical examples of each situation.
Define "sudden infant death syndrome". Briefly describe what we think causes genuine "SIDS", and give a "differential diagnosis".
![]() * Review how to order "arterial blood gases", what you get,
and what they tell you. (Be able to do
this at 3 AM as the only doctor on the ward.) This might be a
good time to look at the
Blood
Gases" handout.
* Review how to order "arterial blood gases", what you get,
and what they tell you. (Be able to do
this at 3 AM as the only doctor on the ward.) This might be a
good time to look at the
Blood
Gases" handout.
Describe the abnormal anatomy and functional problems that every cigaret smoker should expect.
Explain the importance of elastic recoil in keeping respiratory bronchioles patent during exhalation. Explain how this relates to the classic definition of emphysema as "an abnormal, permanent dilatation of part of all of the acinus, with destruction of alveolar walls."
Distinguish the two "classic" types of emphysema, and mention their alleged causes. Tell what we think causes emphysema in cigaret smokers and alpha-1 antitrypsin deficient patients. Tell what a "pink puffer" looks like clinically, and how emphysematous lungs look at autopsy. Describe the complication of "bullous emphysema".
Define "chronic bronchitis" and mention its common causes. Describe the gross and microscopic pathology and the pathophysiology. Be able to define the "Reid Index". Tell what a "blue bloater" looks like clinically, and mention the common organisms that superinfect these patients' lungs.
Define bronchial asthma. Describe its important causes, and distinguish "allergic" and "idiosyncratic" kinds. Describe the common pathophysiology. Tell what you will see at the autopsy of an asthmatic. Mention other causes of wheezing.
Define bronchiectasis. Describe the important causes, the abnormal anatomy, and the typical clinical picture.
Describe the various breathing problems that occur during sleep. Recognize sleep apnea as a common cause of several illnesses.
Describe the normal flora of the lungs in non-smokers and smokers, and recognize the range of micro-organisms that have caused lung infections. Recognize the tremendous clinical importance of lung infections.
Distinguish bronchopneumonia, lobar pneumonia, and pneumonitis. Describe the typical histopathology of lung infections caused by various agents.
List the etiologic agents of lobar pneumonia, the classic stages in its progression, the major complications, and those at risk for each form.
Describe the causes, underlying problems, pathophysiology, and morbid anatomy of bronchopneumonia, aspiration pneumonia, aspiration pneumonitis, legionellosis, pneumocystosis, lung abscess, and viral and mycoplasmal pneumonias. Describe the distinctive features of hantavirus infection and SARS (the 2003 epidemic).
Describe the anatomic pathology, pathophysiology, and clinical picture of the "idiopathic pulmonary fibrosis" family of diseases. Identify the most important members, and mention how we distinguish them and some of the causes.
Define sarcoidosis, and describe a typical sarcoidosis patient. Explain the usual effects of sarcoidosis on the lungs, skin, and eyes. Mention the serious consequences of untreated sarcoidosis. Describe the histology, and give a differential diagnosis for a granuloma found on biopsy. Explain how sarcoidosis causes abnormalities of calcium metabolism. Recognize the Kveim test as of limited usefulness. Tell how to make the diagnosis of sarcoidosis, and how to treat sarcoid patients.
Explain the essential lesion of Goodpasture's disease involving the lung, and mention the clinical picture and diagnostic lab test, and essential treatment. Mention some "related" (?) causes of bleeding from the pulmonary alveoli.
Briefly describe the eosinophilic pneumonias, and the various lipid pneumonias and lipoproteinosis, focusing on their histopathology.
Give the numbers of new cases of lung cancer in U.S. men and women expected this year. Explain how rates are changing, and why. Describe the risk factors for lung cancer, mentioning the importance of cigaret smoking, industrial exposure, radon in the home, and indoor air pollution.
 Explain how and why pathologists subclassify lung cancers.
Be generally familiar with the unsatisfactory WHO-2004 classification:
Explain how and why pathologists subclassify lung cancers.
Be generally familiar with the unsatisfactory WHO-2004 classification:
Describe the important distinctions among these tumors:
Describe what we know about the emerging entity "never-smoker's cancer".
Tell how bronchogenic carcinomas present. Describe the various paraneoplastic syndromes seen with lung cancer, especially the hypercalcemia syndromes and the small cell undifferentiated carcinoma syndromes.
Identify bronchial carcinoid, tell how it looks grossly and microscopically, how to recognize it, and describe its origin and its variants.
List the common problems that affect the larynx or trachea. List the different kinds of pleural effusions, and tell the significance of each. Describe the various kinds of pneumothorax and why they are important. Tell how pleural plaques look and what causes them.
Identify the cell of origin, risk factor, gross and microscopic appearance, and prognosis for malignant mesothelioma.
Mention the basic biology of ciliary dyskinesia syndromes, tell when you would suspect one, and how you would verify it.
As usual, given a gross lung or larynx, or a biopsy of any level of the respiratory tract, recognize any of the lesions exhibited in this section with at least 70% accuracy.
|
|
![]()
Life is not measured by the number of breaths you take, but by the number of moments that take your breath away.NORMAL ANATOMY AND PHYSIOLOGY
-- Attributed to George Carlin
* Click here for Dr. Karius's scheme for normal respiration. You must know this perfectly if you are going to be able to make sense out of lung pathology. Thanks Diane!
* All about the uvula. A human's is much bigger than any other mammal's, and the uvula's job is probably to keep us from getting hoarse while talking (amazing, Yearbook of Path 1994, p. 97 describes its anatomy).
Review the gross anatomy of the respiratory system. Remember that pulmonary artery is anterior to the right mainstem bronchus, and superior to the left mainstem bronchus.
Review the general histology of a BRONCHUS. Bronchi are usually defined to be the airways with cartilage and/or complex glands (precise usage varies). The orders of bronchi end in MEMBRANOUS BRONCHIOLES.
The bronchioles divide further, leading to the TERMINAL BRONCHIOLES, the last division with a continuous non-simple-squamous epithelium (which by this time is probably more likely to be made out of simple-columnar, club-cell rich epithelium -- I prefer "club cell" to Clara-cell as Max Clara was a concentration camp physician).
Remember the importance of the mucociliary elevator provided by this epithelium. The cilia are supposed to be extremely easy to damage and hard to recover, but they almost always are present in my autopsies, even on people who have been very sick for a very long time.
* A strange paper (NEJM 365: 1713, 2011) redefines "terminal bronchioles" entirely by the thickness of their lumens and discovers that folks with emphysema have almost none left. Ignore this for now. Both the editors and I suspect that this really means that the lost elasticity of the alveoli and the thickened mucosas of the real terminal bronchioles explain this.
The portion of lung parenchyma supplied by one terminal bronchiole is called an ACINUS ("respiratory unit", diameter about 7 mm).
From the terminal bronchiole arise several orders of RESPIRATORY BRONCHIOLES, which have part of their walls alveolated and part with pseudostratified respiratory epithelium. These in turn give rise to ALVEOLAR DUCTS and then ATRIA. The ALVEOLI themselves are wide-mouthed sacs that open into all three of these divisions.
Remember that the elastic recoil of the surrounding lung tissue is what keeps the respiratory bronchioles from collapsing when a person starts to exhale.
Schematic diagram of the alveolar wall:

Type I pneumocytes: simple squamous, stretchy, permeable to O2 and CO2, easily injured, do not divide
Type II pneumocytes: reserve cells; cuboidal, granular cytoplasm, produce and process surfactant ("lamellar bodies" in the cytoplasm), divide and flatten to become new type I pneumocytes.
The interstitium contains a few fibroblasts, smooth muscle cells, collagen, elastin.
There are probably no lymphatics in the respiratory membranes themselves, but there are many lymphatics in the fibrous septa between groups of alveoli.
Contrast the alveolar epithelium with that of the bronchi and bronchioles, which includes ciliated, goblet, reserve (basal), Kulchitsky ("K-"), Clara / club, and other types of cells.
Macrophages in the alveoli spaces ("alveolar macrophages") and in the septa ("interstitial macrophages") eat surfactant and most anything else that comes along, in addition to modulating the immune response locally.
You are already well-aware that the main pulmonary arterial circulation runs with the bronchi and bronchioles, and the the pulmonary veins run separately. Remember that the lung receives a dual blood supply, from the pulmonary (thinner walls) and bronchial arteries (thicker walls - why?). The bronchial arterial supply is one of the first to stop in low-output states (left heart failure, shock; why?)
Bronchial lymphoid tissue (BALT) shouldn't be present until a baby is a few months old. Lymphocyte clusters in a newborn's large airways means infection. Smokers have much more "BALT" than do non-smokers.
Remember blood PaCO2 is almost entirely a function of overall alveolar ventilation.
Remember blood PaO2 is a function of the quality of ventilation-perfusion matching, alveolar septal thickening (if excessive), and overall ventilation. And remember that lung structures are generally much less permeable to O2 than to CO2 (a fact that becomes important only in disease.)
Don't forget either that blood with PaO2 of 65 torr is carrying almost as much oxygen as blood with PaO2 of 100 torr or higher, because most oxygen is bound to hemoglobin (recall the hemoglobin-oxygen dissociation curve....) It's when PaO2 drops to around 60 that you start getting into trouble, because the hemoglobin stops binding oxygen as well as it should.
Cyanosis appears when the arterial blood contains 5 gm % or more of unoxygenated hemoglobin.
All of the first group of diseases on the handout (except pulmonary thromboemboli) are PATTERNS of lung injury, each of which has many different causes.
* BIRTH DEFECTS involving the lungs are uncommon. If the diaphragm did not form properly and the abdominal contents are in the chest cavity, or there was insufficient amniotic fluid ("oligohydramnios") or the ribs or the large airways did not develop, there may not be sufficient lung tissue to allow life outside the womb.
PULMONARY CONGESTION AND EDEMA
{37956} pulmonary edema gestalt
PULMONARY EDEMA classically results from the same factors that cause edema in the rest of the body. These are:
1. Increased venous hydrostatic pressure (left-sided heart failure)
2. Fluid overload (renal failure, iatrogenic, etc.)
3. Decreased albumin in the blood (liver disease, nephrotic syndrome, poor nutrition, etc.) -- theoretical concern but is probably only a contributing factor in severe disease where there are other problems.
4. Lymphatic obstruction (cancer, etc.)
5. Endothelial damage (i.e., the pneumonias; most striking is the hantavirus that first struck in the US southwest; also remember poisons, especially phosgene -- a hay-scented liquid which could easily be used by terrorists)
6. Getting strangled / physically asphyxiated (negative pressure)
You have to learn these causes of pulmonary edema for which the mechanisms are not well understood:
7. Acute CNS injury (* perhaps reflex constriction of the pulmonary veins?)
8. Opiate overdose (* perhaps reflex constriction of the pulmonary veins?)
9. Re-expansion after placement of a chest tube
10. Exposure to high altitudes (unacclimatized people; review of the still-puzzling
high-altitude illnesses -- both pulmonary edema and cerebral edema -- Chest 134: 402, 2008;
Am. Fam. Phys. 82: 1103, 2010; NEJM 368: 2294, 2013.)
However, this can't be the full story, because of the pharmacology (Br. Med. J. 321: 267, 2000). Dexamethasone (the familiar glucocorticoid) apparently prevents acute hypoxia from making endothelial cells more permeable, while how acetazolamide (a carbonic anhydrase inhibitor) works is anybody's guess. The finding that a beta-agonist helps is attributed to increased active transport of sodium from the alveolar fluid into the bloodstream (NEJM 346: 1631, 2002). |

|
* Populations that inhabit high mountains seem to survive only by making huge amounts of nitric oxide, which you can measure on their breath. See Nature 414: 411, 2001. Natural selection or physiologic adaptation? We don't yet know. It's finally documented that mountain dwellers have greatly expanded chest cavities: Resp. Phys. Neuro. 132: 223, 2002. Around 140 million people live above 8200 feet / 2500 meters, and many of these develop polycythemia and/or cor pulmonale eventually as a result (Chest 137(S6): 13-S, 2010).
In each case, when the capacity of the lymphatics to drain the interstitial fluid is exceeded, INTERSTITIAL EDEMA develops, with loss of lung compliance and a barrier to oxygen exchange (alveolar-capillary block.)
As the interstitial pressure rises still further, the tight junctions between the alveolar epithelial cells open and fluid pours into the alveolar spaces, causing ALVEOLAR EDEMA and stopping ventilation.
Alveolar edema fluid is a good culture medium for bacteria. Secondary pneumonia is common.
When you hear CRACKLES (rales) through your stethoscope, you're hearing the little air bubbles in the alveoli. Your patient has alveolar pulmonary edema, and you must figure out why. Fix it, and the edema fluid will return to the heart by way of the abundant lymphatics.
![]() {10145} pulmonary edema (just enough protein
content to stain...)
{10145} pulmonary edema (just enough protein
content to stain...)
{11666} pulmonary edema
|
|
|
PULMONARY CONGESTION of course results from increased pulmonary venous hydrostatic pressure, typically from left-sided heart failure especially mitral valve disease.
If marked or longstanding, microhemorrhages, fibrosis, and iron pigmentation ("brown induration") occur in the lungs. (Hemosiderin-laden macrophages are called "heart-failure cells.")
|
|
|
PULMONARY EMBOLIZATION ("embolism") AND INFARCTION (NEJM 358: 1209, 2008; Lancet 363: 1295, 2004; the enormity of the deep-vein-thrombosis problem and what may lie ahead Lancet 379: 1835, 2012.)
Pulmonary thromboemboli are very common (and still under-diagnosed -- see Arch. Int. Med. 148: 1345, 1988, also still good). Thrombi in unoperated pulmonary arteries are almost always emboli.
Most originate in the deep veins of the legs; they may also come from the pelvic veins, rarely cerebral dural sinuses or elsewhere.
Thrombosis in a leg vein can be uncomfortable ("thrombophlebitis"), but is most often asymptomatic. As a junior clerk, you will compare calf circumferences, check Homan's sign (be careful you don't break the thrombus off), etc.
The majority of pulmonary thromboemboli do no harm and eventually organize or lyse; many are fatal.
Pathologists report pulmonary emboli in 8-25% of autopsies on hospital patients. But in 3-5% of autopsies (figures vary), the embolus is the fatal event, and estimates of the number of U.S. deaths from pulmonary emboli are in the 50,000-180,000 range. (JAMA 309: 171, 2013; without an autopsy or angiogram, the clinicians will often tell the family, "heart attack".)
Virchow's triad. Typical settings for deep vein thrombosis and pulmonary thromboemboli include:
2. hypercoagulable states
3. damaged endothelium
Nobody knows why -- babies very seldom get or die of pulmonary thromboemboli: Arch. Path. Lab. Med. 114: 142, 1990.
Pulmonary thromboemboli cause several types of problems.
Pulmonary infarcts are peripheral and hemorrhagic (and can even cause hemolytic jaundice if the patient survives. Rising bilirubin and rising LDH-3 -- think of a pulmonary infarct.) Listen for a friction rub; look for fibrin on the pleural surface at autopsy (why?). They can be distinguished best from regular intrapulmonary hemorrhages by the necrosis. Most will have the classic "wedge" shape.
Infected thromboemboli can cause "septic infarcts". These may become lung abscesses.
Any patient with sudden anxiety, chest pain, dyspnea, cough, hemoptysis or sudden death should make you think of pulmonary emboli. You'll learn how to make the diagnosis while on rotations -- but don't get over-confident; pulmonary embolization is the most-often-missed diagnosis in the hospital (Arch. Path. Lab. Med. 129: 201, 2005).
Fibrin D-dimer is sensitive, but not specific: Arch. Path. Lab. Med. 123: 235, 1999. Some folks consider this the best screening test: Lancet 363: 1295, 2004. Others say to continue the investigation into any patient who really seems to have a pulmonary embolus, even if the d-dimer is normal (Chest 134: 789, 2008). Today, unless the patient seems very sick indeed, d-dimer is good enough to rule out pulmonary embolus (NEJM 363: 266, 2010; Am. J. Clin. Path. 132: 326, 2009; setting the cutoff higher in older folks is helpful BMJ 344: e2985, 2012).
As we have already mentioned, pulmonary emboli organize into fibrous bands that remain in the lung for the rest of the person's life (Arch. Path. Lab. Med. 123: 170, 1999).
{29052} pulmonary embolus, ancient, organized and turned into fibrous bands
|
Australian Pathology Museum High-tech gross photos
|
|
|
Other causes of LUNG INFARCTION (immune or infection vasculitis, other things embolizing) are rare but not unheard-of (Chest 127: 1178, 2005).
FAT EMBOLIZATION remains mysterious and probaby underdiagnosed. You're familiar with this dread complication of a bony fracture. Today's pathologists make the diagnosis during life by finding fat in the cells obtained by pulmonary lavage. Update J. Trauma 71: 312, 2011.
The deadly CHEST SYDROME in sickle cell anemia may also result (at least sometimes) from fat embolization after a bony infarct (NEJM 342: 1904, 2000). The same picture can appear in sicklers with bacterial or viral infection or thromboembolization; often no cause is found. I suspect that cells sickling in the presence of reduced oxygen tension is part of the cause.

PULMONARY HYPERTENSION
|
|
Generally analogous to systemic arterial hypertension, pulmonary hypertension also results from a variety of causes.
1. Left-sided heart failure (especially mitral valve stenosis)
2. Increased blood flow into the pulmonary arteries (i.e., congenital cardiac malformations with left-to-right shunts, or after resection of a lung)
3. Increased pulmonary vascular resistance from any cause.
NOTE: This is important. The normal response to hypoxia in the alveoli ("hypoxic pulmonary vasoconstriction"; formerly the "hypoxic vascular response") causes constriction of arterioles supplying underoxygenated alveoli (probably because alveolar epithelium produces less nitric oxide when there is insufficient oxygen). If all the alveoli are underoxygenated, all the arterioles constrict, and the pulmonary arterial blood pressure has to rise. To this day, nobody knows which mediator produces the response. Update Ann. Thor. Surg. 78: 360, 2004.
Treating it with calcium channel blockers (helps, no panacea): NEJM 327: 76, 1992; nitric oxide and continuous intravenous prostacyclin are new remedies (Chest 119: 970, 2001; Am. J. Card. 75: 51A, 1995; big review); also Lancet 352: 719, 1998; NEJM 338: 273, 1998; aerosolized iloprost (prostacyclin analogue: NEJM 342: 1866, 2000; reversal in a mouse model using a serine elastase inhibitor: Nat. Med. 6: 698, 2000.
* Bosentan, the endothelin receptor antagonist, seems to help "idiopathic" primary pulmonary hypertension: Lancet 358: 1119, 2001; NEJM 346: 896, 2002. Similar medicines have been approved since, and sildenafil "Viagra" was approved in 2005.
The prognosis is still poor, with most of these patients dying of their illness (most often right heart failure or sudden death Am. J. Resp. Crit. Care Med. 188: 365, 2013).
The drug sildenafil, better-known as Viagra, causes the smooth muscles of the little pulmonary arteries to relax, thereby relieving pulmonary hypertension. This is already in use for idiopathic pulmonary hypertension (NEJM 353: 2148, 2005; South. Med. J. 99: 880, 2006, many others); watch it for fibrosing alveolitis (Chest 131: 897, 2007) and other lung diseases with vascular damage.
NOTE: You'll frequently hear, "This patient is disabled because of pulmonary hypertension" or "This patient has right sided heart failure due to pulmonary hypertension". Of course, the real problem is increased pulmonary vascular resistance from thickened and narrowed arterioles.
Sustained pulmonary hypertension results in changes in the anatomy of the pulmonary arteries and arterioles, some of which are irreversible.
The principal molecular suspect is endothelin 1, derived from damaged endothelium. It is a potent vasoconstrictor, and makes smooth muscle proliferate. See Ann. Int. Med. 114: 213, 1991; NEJM 328: 1732, 1993.
* By contrast, the ability of the lung endothelium to produce nitric oxide ("endothelium derived relaxation factor", EDRF), a potent vasodilator, is much diminished in emphysema. There's also maybe a lack of prostacyclin. Nobody really understands this.
Unlike in "benign" systemic hypertension, some of these changes clearly contribute to the ongoing process.
Lesions include:
Main arteries: atherosclerosis (never severe)
Small arteries: hyalinization ("hyaline arteriolar sclerosis"), intimal onion-skinning ("hyperplastic arteriolar sclerosis"), extra elastic, extra muscle, fibrinoid.
Arterioles: muscle appears (should be none), plexiform and angiomatoid lesions (bad sign)
* Future pathologists: Don't overcall the sperrarteries, tubes of longitudinally-oriented smooth muscle with almost no lumens that are connected to the bronchial arteries in some normal lungs, as "pulmonary hypertension."
![]() Plexiform lesions
Plexiform lesions
Lung pathology series
Dr. Warnock's Collection
Pulmonary hypertension (especially, pulmonary hypertension due to increased pulmonary vascular resistance) is common and under-appreciated.
A very common mechanism of sudden death in generalized lung disease is rhythm disturbance arising in the strained right ventricle.
And no matter what the underlying disease, the pulmonary vascular resistance is likely to determine the patient's exercise tolerance ("quality of life"). This commonly takes precedence over the "respiratory function tests" of spirometrists (Chest 92: 387, 1987).
Using nitric oxide to control pulmonary hypertension in ARDS seems natural and physiologic: NEJM 328: 399, 1993. Update: Chest 115: 1407, 1999. It is now a mainstream, though expensive, treatment for pulmonary hypertension from most causes.
Future pathologists only: Here's a simplification of the standard grading system for pulmonary hypertension vascular lesions:
|
|
|
ACUTE / ADULT RESPIRATORY DISTRESS SYNDROME (ARDS -- update Lancet 369: 1553, 2007; Am. Fam. Phys. 85: 352, 2012)
|
This very common, deadly, and expensive problem results from anything that severely injures the type I pneumocytes and capillary endothelial cells throughout the lungs.
Clinicians distinguish "pulmonary ARDS" (caused by direct lung injury) from "extrapulmonary ARDS" (a remote effect of injury elsewhere). The outcome depends on the severity of the illness rather than the type (Chest 133: 1463, 2008).
Historically, there have been at least 150,000 ARDS cases in the U.S. every year (as an autopsy pathologist, I'd say this about right).
ARDS first came to be recognized during the Vietnam War.
It was called "Da Nang lung". Originally there was concern about biological warfare. But it soon became clear that the pulmonary changes were remote effects of injuries that soldiers in previous conflicts did not survive. |

Siege of Da Nang |
ARDS has many causes. These include...
It's most common with blood products that have the most donor antibodies (fresh frozen plasma, platelets.) * Neutrophils' binding to platelets as a mechanism in "TRALI" and perhaps many other diseases: Nat. Med. 15: 384, 2009.
* Mouse model for TRALI: J. Clin. Inv. 119: 3450, 2009. Depleting / inactivating platelets seems to prevent it.
* Scanning and transmission electron microscopy of TRALI -- is the damage done by cholesterol needles? Am. J. Clin. Path.140: 170, 2013.
Probably the mechanism is two-stage. First, in serious illness, there are more neutrophils in the lung microvasculature. Second, when antibodies in the transfused blood product bind to their HLA-1 molecules, they are damaged and their contents in turn damage the lung. This is why we prefer to use plasma from untransfused male donors when possible -- antibodies against other people's HLA antigens are usually acquired in pregnancy.
Mechanisms of injury are complex, with free radicals, complement, enzymes from marginated polys (can't be the whole story, since the neutropenic can and do get ARDS), microthrombi, aggregation of polys, "shock toxins", and many other ideas.
One major experimental model involves reperfusion of an animal's hindlimb rendered ischemic for a considerable time (J. Traum. 31: 760, 1991).
Neutrophils are very abundant in the alveolar capillaries especially in early shock lung, and you can make the diagnosis this way at autopsy.
Interleukin-8, a great neutrophil attractant, abounds in fluid from ARDS lungs. We don't know why, but this probably has a lot to do with the problem.
Biopsy is often helpful to establish an underlying diagnosis even though these people are very sick (Chest 125: 197, 2004. Here are some tips for the biopsy pathologist for guessing the cause of ARDS:
Whatever causes the injury, the result pretty much the same:
When the alveolar cells are injured fluid leaks into the interstitial spaces and alveolar air spaces -- this is PULMONARY EDEMA.
Later, with cell necrosis, FIBRIN is released into the alveoli, producing HYALINE MEMBRANES. Of course there is LOSS OF SURFACTANT, so many alveoli COLLAPSE.
During this early stage, the patient is very tachypneic and dyspneic, but the chest x-ray looks normal. (Why?)
NOTE: In "respiratory distress syndrome" or "hyaline membrane disease" of low-birth-weight infants, the lack of surfactant is one primary problem, though not the only one. In ARDS, surfactant is decreased secondary to diffuse alveolar damage.
* Surfactant aerosol-replacement flops for ARDS: NEJM 334: 1448, 1996.
As type I pneumocytes are destroyed, TYPE II PNEUMOCYTES DIVIDE to replace them ("regenerative epithelial hyperplasia" or CUBOIDALIZATION of alveolar epithelium.) Of course, they are not so permeable to oxygen as the healthy type I cells.
FIBROSIS ensues as the intra-alveolar hyaline membranes and the interstitial exudate organize (Am. J. Path. 126: 171, 1987).
Of course, fibrotic lung (from any cause) is prone to develop bacterial infections (bronchopneumonia), since it is harder to mobilize the exudate and perhaps the neutrophils must travel farther in pursuit of the microbes.
Even where the alveoli remain ventilated, these factors combine to cause POOR PULMONARY COMPLIANCE plus POOR RESPONSE TO OXYGEN THERAPY ("alveolar-capillary block" in damaged alveolar walls; I'm inclined to believe in this.) Further, the PULMONARY VASCULAR BED is progressively obliterated.
|
|
{06359} ARDS
The outcome depends on whether the patient can be supported and the underlying problem successfully treated before fibrosis becomes extensive.
Nobody knows exactly why (maybe relieving the weight pressing on the pulmonary veins), but putting the patient prone often helps oxygenation in the short-term (and maybe long-term survival: NEJM 345: 568, 2001; J. Trauma 72: 1634, 2012); NEJM 368:2159, 2013.
About 50% of patients with ARDS die from it. Long, agonizing periods on the ventilator are fairly common. The ARDS autopsy and clinical correlations: Ann. Int. Med. 141: 440, 2004.
* Today, there is considerable interest in factors that enhance regeneration of the pulmonary endothelial cells (J. Clin. Inv. 116: 2316, 2006).
Even more interesting, the prevalence of ARDS decreased dramatically between 2000 and 2007, especially in trauma patients (J. Trauma 63: 1, 2007). When one tries to focus on what we've been calling ARDS, and not just the problems of the immediate post-injury period like contusions or fluid problems, it turns out that with today's more conservative therapy (less transfusion, lower tidal volumes, lower breathing-machine pressures, less overloading with fluids -- all things that you'd think would damage the alveolar membranes), the rate of bad ARDS has been cut about 75%.
| * With more patients surviving ARDS nowadays, there is considerable interest in the quality of these survivals. There is often brain damage sufficient to impair the quality of life (AJRCCM 171: 340, 2005), post-traumatic stress disorder (Am. J. Psych. 161: 45, 2004), and of course impaired lung function (Chest 123: 845, 2003; NEJM 348: 683, 2003). Update NEJM 364: 1293, 2011. |
 Anakin Skywalker "Darth Vader" ARDS survivor |
NEONATAL RESPIRATORY DISTRESS SYNDROME ("hyaline membrane disease", HMD, RDS, etc.)
This is the common cause of respiratory distress in premature infants (usually 1500 gm or under), beginning a few hours after birth.
In addition to prematurity, risk factors include maternal diabetes, caesarean sections, premature rupture of the membranes. Heroin addiction or glucocorticoid administration in the mother protects from RDS.
The pathophysiology is lack of surfactant plus high permeability of the immature pulmonary epithelium. A few air spaces are hyperinflated, the rest of the lung is collapsed.
The principal lesion is probably necrosis of the respiratory epithelial cells; why this happens to premature infants is unclear but probably has something to do with their being unable to tolerate even normal amounts of inspired oxygen.
Surfactant is also deficient in these patients and this accounts for part of the problem. Surfactant -- dipalmitoyl lecithin -- is the stuff that keeps the alveoli uniform in size. Recall the "two balloons on joining pipes" model in physiology.
In RDS, hyaline membranes line the open alveoli (mostly along the respiratory bronchioles). As in ARDS, they result from plasma proteins exuding through the alveolar walls. (* These kids usually have patent ductus arteriosus with marked left-to-right shunting, which greatly exacerbates this problem.)
As in ARDS, the presence of hyaline membranes is both a marker for alveolar membrane injury, and a further barrier to gas exchange.
{11427} hyaline membrane disease, newborn
{20014} hyaline membrane disease, newborn
{20015} hyaline membrane disease, newborn
|
|
To test for maturity of an unborn child's lungs, check the lecithin-to-sphingomyelin (L/S) ratio in amniotic fluid obtained by amniocentesis -- should be 1.5 or more.
In the old days, treatment was limited to ventilatory support and oxygen, plus drugs to dilate the pulmonary arterioles.
During the 1990s, preparations of surfactant became available that could be administered to the child at birth, before the first breath. They helped, but were not a panacea.
A significant number of low-birth-weight kids do suffer brain damage from hyaline membrane disease hypoxemia: NEJM 325: 276, 1991.
* The extracorporeal membrane oxygenator (ECMO) is a modified heart-lung machine for preemies. The economic-ethical nightmares still happen.
Big doses of oxygen, plus barotrauma and whatever else happens in the premature lung, cause further alveolar damage, resulting in fibrosis and/or failed development of alveoli, which is given the unfortunate name of BRONCHOPULMONARY DYSPLASIA (it is neither incipient cancer, nor a birth defect). Today it is better to call it CHRONIC LUNG DISEASE OF PREMATURITY though you'll see both terms. There's less of it now than in the old days because of surfactant replacement and less oxygen adminstration, but whether extremely low-birth-weight children are given surfactant, CPAP, and/or intubated, it's still all too common (NEJM 358: 1700, 2008; NEJM 362: 1970, 2010).
* The "forme fruste" may be a tendency in survivors of neonatal hyaline membrane disease to develop pulmonary hypertension more easily later in life (Am. J. Path. 178: 201, 2011).
![]() Bronchopulmonary "dysplasia"
Bronchopulmonary "dysplasia"
WebPath Photo
Historically, the outcome was usually bad for lung and brain long-term (Pediatrics 77: 345, 1986). Many of these babies spent months or years on ventilators before finally dying.
Today, fibrosis is less common (perhaps thanks to less aggressive ventilator therapy?), and "bronchopulmonary dysplasia" has been redefined as oxygen dependency at 36 weeks -- perhaps explaining why the prognosis is better. Children coming to autopsy often have too few alveoli, the ones they have of course being overdistended. Update on the pathology: Am. J. Resp. Crit. Care Med. 181: 1093, 2010.
* Other problems of preemies: patent ductus arteriosus, meconium aspiration, necrotizing enterocolitis, cerebral hemorrhages from the germinal zone of the subependymal plate, preemie retinopathy (oxygen in particular ...keeping the oxygenation at 85-89% rather than 91-95% decreases retinopathy but increases overall mortality: NEJM 362: 1959, 2010).
* In genetic absence of surfactant, babies born at term are unable to inflate their lungs. The disease is fatal: NEJM 350: 1296, 2004.
ATELECTASIS
|
|
|
Collapse (or incomplete expansion) of pulmonary acini from any cause.
OBSTRUCTIVE ATELECTASIS ("absorption atelectasis") results from non-ventilation of alveoli that are still perfused; the alveolar gas is carried away by the bloodstream.
Seen distal to tumors, foreign bodies, mucus blobs, post-surgical
discomfort preventing cough, enlarged
hilar nodes (cancer, TB![]() -- producing "the right middle lobe
syndrome") etc. Before antibiotics, this was a setup for
bronchiectasis.
-- producing "the right middle lobe
syndrome") etc. Before antibiotics, this was a setup for
bronchiectasis.
If an airway is obstructed, expect that over time the alveoli will fill with surfactant, which will mostly be engulved by macrophages. This is the "obstructive / postobstructive pneumonia" or "golden pneumonia", and often is the first x-ray sign of lung cancer.
COMPRESSIVE ATELECTASIS results from something in the pleural cavity (blood, exudate, tumor, air.)
If extensive and unilateral, obstructive and compressive atelectasis can be distinguished by the direction in which the mediastinum is shifted on x-ray (think about it).
A deficiency of surfactant produces "patchy atelectasis" in both hyaline membrane disease ("fetal atelectasis" -- * a term also used for lungs of stillborns who never breathed) and ARDS (see below.)
![]() Atelectasis
Atelectasis
This was from a hemothorax
WebPath
{10228} atelectasis (left lung of a baby whose left bronchus failed to form)
* ROUND ATELECTASIS ("folded lung", "shrinking pleuritis"): a vanishing coin lesion, resulting when normal lung parenchyma is crunched into a little ball beneath a shrinking pleural scar. Familiar to radiologists, sometimes operated for diagnosis.
SUDDEN INFANT DEATH SYNDROME ("SIDS", "crib death", "cot death", "continues to be the leading cuase of death for infants aged between 1 month and 1 year in developed countries" Lancet 370: 1578, 2007; NEJM 361: 496, 2009; "the seventh leading cause of years of potential life lost before age 65, commensurate with AIDS" -- MMWR 37: 644, 1988, etc., etc.). The following will upset you.
Sudden death in an apparently healthy baby, less than one year old, with no explanation even after autopsy. Sources in the 1970's and 1980's claimed that this affected 2-3 per 1000 live births, "the single most common cause of infant death" (Baby Robbins, etc., etc.) -- slightly more common than baby death due to birth defects.
There is no question that genuine SIDS cases exist, i.e., some babies do die from disturbed pathophysiology without any environmental or anatomic explanation. A few causes are known (i.e., channelopathy) or suspected (not being able to protect the airway while sleeping; actual problems with respiratory drive; these must be rare). BUT TO UNDERSTAND "SUDDEN INFANT DEATH", WE FIRST NEED TO SORT OUT DEATHS THAT AREN'T "SIDS" AT ALL. Thankfully, this is now happening.
During the 1960's, mostly because of the militancy of a single pediatrician-activist, certain anti-common-sense ideas about the sudden deaths of infants were suddenly and uncritically accepted, both by the medical community and by the public. Looking back, this had a lot to do with the "feel good / no blame / flower power" mentality of the times. It was dogma that:
That this was wishful thinking should have been obvious, even at the time.
The classical autopsy finding in SIDS is petechiae over the thymus, lungs, and heart, without other abnormalities... or with nasal hemorrhages too. And this is exactly what you'd expect to find in a baby who has been suffocated -- accidentally, intentionally, on the bedclothes, or against the mattress (JAMA 263: 2865, 1990; Arch. Dis. Child. 85: 116, 2001).
And it was already well-known that "higher rates are encountered in many developing countries" (Baby Robbins), and especially among the underclass in industrial countries (Practitioner 232: 577, 1988; Arch. Dis. Child 65: 830, 1990; risk for First Americans correlates with underclass risk factors rather than genes J. Ped. 121: 242, 1992; also & JAMA 288: 2717, 2002; underclass status still correlates very strongly with "SIDS" risk in the USA, Australia, New Zealand, and South Africa NEJM 361: 496, 2009.)
Some classic reported SIDS death rates:
0.04%... Rich New York suburbanites
0.4%... Poor New York slum dwellers (both from NEJM 315: 100 & 126, 1986)
0.35%... North Plains Indian reservations (JAMA, above)
2% (!!) Children of British criminal offenders (Arch. Dis. Child. 62: 146, 1987.)
near zero... Hong Kong -- attributed to zero privacy and many helpers for mothers, no junk in the cribs, "nobody lets her baby sleep prone", "no unwanted babies": Lancet 2: 1346, 1985; Br. Med. J. 298: 721, 1989.
It is now obvious that many (if not most) deaths signed out as "SIDS" during the decades of ignorance were the results of negligence or abuse by family members. This was substantiated in the 1980's by careful death-scene investigators (filicide Arch. Dis. Child. 60: 505, 1985; NEJM, above; Lancet 1: 313, 1986; Lancet 1: 199, 1989; J. Ped. 117: 351, 1990.)
There was never any basis for believing that overlying cannot kill a baby. (King Solomon's judgement; Yeats's Moll Magee; most cultures historically have simply accepted that overlying kills babies.) See also Am. J. Forensic Med. & Path. (8): 256, 1987; when we stop deceiving ourselves, overlying turns out to be common: Am. J. Dis. Child. 146: 968, 1992.) Sharing a bed with a parent as a risk for SIDS: Br. Med. J. 319: 1457, 1999; Arch. Path. Lab. Med. 126 343, 2002; J. Ped. 147: 32, 2005; Arch. Dis. Child. 88: 1058, 2003; Lancet 363: 185, 2004; Arch. Dis. Child 91: 318, 2006; Lancet 314: 367, 2006 ("Although the reason for the rise in deaths when a parent sleeps with their infant on a sofa are still unclear, we strongly recommend that parents avoid this sleeping arrangement."); J. Ped. 160: 44, 2012. Sofas are worse and so is sleeping with Mom when she is drunk or on drugs (go figure; Arch. Dis. Child. 88: 112, 2003). There are still plenty of people in the USA -- mostly breast-feeding militants and a few "SIDS activists" -- who promote co-sleeping with infants; some talk about "safety precautions". Perhaps it's thanks to these folks that in progressive Massachusetts, 37.9% of "sudden infant death syndrome" victims were co-sleeping (up from 19.2% in the pre-"Back to Sleep" era -- Pediatrics 129: 630, 2012).
Drug use by Mom ("during pregnancy", and of course most likely continuing after) is a very powerful risk factor for SIDS, especially when the drug is heroin or methadone, and when other risk factors (poverty, smoking, and so forth) are controlled-for (J. Ped. 123: 120, 1993). A classic observation is a higher incidence (+ 60% or so) of "SIDS" on weekends (Aust. N.Z. J. Med. 18: 861, 1988; still true Arch. Dis. Child. 89: 670, 2004), when Mom and Dad/current boyfriend are more likely to be drunk or stoned. This is every bit as true today as before the "back to sleep" campaign (Arch. Dis. Child. 89: 670, 2004 -- no surprise). And children of schizophrenic mothers have around five times the risk of "SIDS" (Arch. Gen. Psych. 58: 674, 2001).
The fact that "SIDS is more than ten times more frequent if a sibling has already died of SIDS" (Arch. Dis. Child. 64: 179, 1989) hardly proves that "bad genes" are responsible. (No adoption studies are available yet....) The fact that SIDS rates are more than double for very young parents (J. Ped. 116: 520, 1990) hardly proves that some mysterious intra-uterine factor is involved. Nor does the fact that SIDS rates are double if the parents smoke (Am. J. Pub. Health 80: 29, 1990) prove that the cause is subtle irritation of the airways.
And the fact that twins used to die of "SIDS" at exactly the same time invites an obvious conclusion (Am. J. Forens. Med. Path. 10: 200, 1989; AJFMP 19: 195, 1998). The fact that this no longer happens (Arch. Ped. Adol. Med. 153: 736, 1999) just says to me that pathologists are now recognizing double infanticides.
The traditional wisdom that "SIDS" typically follows a minor illness now seems to be unfounded (Lancet 300: 1237, 1990). The claims about immunization being a risk factor are clearly untrue; statistically, children actually seem protected (Br. Med. J. 322: 822, 2001; the role of coincidence Pediatrics 115: e643, 2005).
And the familiar junk-science claim by breast-feeding militants that SIDS is due to bottle feeding simply isn't true: Br. Med. J. 310: 88, 1995. At best, the relationship is statistical, and it's weak. However, the SIDS organizations still tell people that breast-feeding protects children from SIDS, which must put a terrible burden of guilt on people whose babies died of the real thing.
Possibly a few infants do die from failure of respiratory drive during sleep. "Near-miss" apnea and related respiratory rhythm disturbances are demonstrable in some siblings, though the findings are notoriously un-reproducible.
* I was impressed only by Hum. Genet. 73: 39, 1986 -- a family with very little myelin in the respiratory centers of their medullas). Since this hasn't been replicated, I wonder about the neuropathologist's myelin stain. Finding hypoplasia of the arcuate (CO2 sensor) nucleus of the medulla in a subset of SIDS is interesting: J. Neuropath. 51: 394, 1992; I'd bet these brains are those that don't respond to having the mouth and nose up against the mattress. An Italian group claims to have found "frequent alterations, mainly cogenital, of the autonomic nervous system" (Am. J. Clin. Path. 124: 259, 2005; also J. Clin. Path. 58: 77, 2005). More credible is a big study from Harvard (JAMA 296: 2124, 2006 and now JAMA 303: 430, 2010) on the serotoninergic neurons in the medulla in SIDS. However, most pathologists don't do microscopy on a brain of a SIDS victim unless it's grossly abnormal, and this is probably right (J. Clin. Path. 65: 257, 2012; just take heart, lung, liver and kidney Am. J. Clin. Path. 65: 58, 2012). Stay tuned.
Otherwise, "near-miss apnea seen in siblings" is famously non-lethal: Science 264: 197, 1994. I suspect that, in most cases, "near-miss SIDS" is Cheyne-Stokes respirations (as a junior med student sharing a call room, I was told I do this while asleep) or transient obstructive sleep apnea.
It is now obvious that some cases of SIDS result from laying babies prone (Pediatrics 93: 814, 1994; Pediatrics 105: 650 2000; JAMA 273: 783, 1995, lots more). Most plausibly, sleeping babies ("perhaps with a genetic predisposition") might simply fail to move when their faces lie flat against mattress, and smother in this way.... (gee whiz) ... or rebreathe carbon dioxide, which is pretty much the same thing (Am. J. Dis. Child. 147: 642, 1993; J. Ped. 122: 874, 1993). This is consistent with some studies that strongly suggest prolonged hypoxia has occurred prior to death in many SIDS cases (Pediatrics 87: 306, 1991, others); nowadays, "near-miss SIDS" is even being observed in the delivery room when the exhausted mother is encouraged to "cuddle and try to breast-feed Baby right now (Pediatrics 127: e869, 2011).
There has been about a 50% reduction in "SIDS" in countries where there's been a campaign to get parents not to place their babies prone. The impact is extremely obvious (Lancet 363: 185, 2004). In the U.S., where "SIDS activists" insisted for two decades that "SIDS is not your fault and a child cannot smother against the mattress", the campaign was delayed. Figure out yourself how many children died as a result. "Suffocated prone: the iatrogenic tragedy of SIDS": Am. J. Pub. Health 90: 527, 2000.
There's some interesting work on facial morphology in these kids -- their upper and lower jaws seem to be, on average, farther back, and perhaps this makes it easier for them to smother on their mattresses (Lancet 317: 293, 1998). The relationship with obstructive sleep apnea -- which this group claims is more common in SIDS families -- is going to take more study to work out.
![]() SIDS
SIDS
Instructional material
WebPath Photo
When you are working in the neonatal intensive care unit, you will frequently see preemies that have trouble breathing or even stop breathing ("apnea of prematurity") or suffer cardiac rhythm disturbances. That this has nothing to do with SIDS was established long ago (Pediatrics 77: 811, 1986; Pediatrics 78: 787, 1986); today it's treated with caffeine and this seems safe (NEJM 357: 1893, 2007.
Public awareness of "SIDS" resulted in a huge "apnea monitor" industry (racket, or "expensive cover-your-butt stuff", as almost everybody will tell you nowadays). For over two decades, the people who really know about SIDS laughed at the idea that monitors save lives (see, for example, Chest 91: 898, 1987; Can. Med. Assoc. J. 140: 1072, 1989), but they did keep parents from staying up all night watching their babies breathe. "Consensus document" on home monitoring: Pediatrics 79: 292, 1987; noncompliance or worse: Am. J. Dis. Child. 142: 1037, 1988. By 1994, the idea that apnea is an important cause of SIDS is dismissed by almost the entire scientific community, though still believed by physicians (Science 264: 197, 1994); however it's still routinely prescribed (Arch. Dis. Child. 84: 270, 2001). To clinch the matter, Waneta E. Hoyte, the mother whose "tragic story" led to the paper (Ped. 50, 646, 1972) that spawned the apnea monitor business confessed in 1994 to having smothered her five children. ("Their screaming made her feel useless": Ped. 93: 944, 1994).
Some "clever" parents learn to stop their kid from crying by smothering it into unconsciousness; this will load the lungs with hemosiderin-laden macrophages (why?). Eventually, these kids are likely to die. Pathologists are now routinely signing "SIDS" cases with lots of these macrophages as "manner of death undetermined" (J. Clin. Path. 52: 581, 1999) and staining all infant lungs to see hemosiderin (Am. J. For. Med. Path. 23: 360, 2002).
The British estimate that 1 in every 10 SIDS cases is a covert homicide (Arch. Dis. Child. 89: 443, 2004); whatever you think of the estimate, common-sense tells you when to suspect murder. SIDS "research" and political agendas: Arch. Dis. Child 88: 1085, 2003.
Most recently, the British have questioned whether "SIDS" EVER really occurs. If you accept that a child can die from second-hand smoke (I'm undecided), and recognize accidental asphyxiation (fifty percent of the kids who died were co-sleeping -- I'm not making this up), "unexplainable deaths" almost disappear (BMJ 336: 302, 2008). The American Academy of Pediatrics, in its massive "policy statement" on SIDS (great reading) advised against co-sleeping in 2005 (Pediatrics 116: 1245, 2005). In the USA, there are still (2008) pro-co-sleeping militants "fighting the crib industry".
And famously, the British went too far in convicting Angela Cannings and Sally Clark on bad medical testimony. Roy Meadow MD, famous for his 1977 work on "Munchausen's by proxy" and very popular in child-protection circles, was the physician responsible for both fiascoes. He used the familiar lawyer's probability fallacy in court ("What are the odds against....?") In doing so, he also assumed that all true SIDS struck at random and overlooked the possibility of a genetic tendency. ("Did you ever hear of the channelopathies, Roy?") The courts eventually found him guilty of grave professional misconduct. Roy was stripped for a while of his medical license, which was probably right. I felt these cases showed the need for forensic experts focused on criminal defense.
Before you write "SIDS" on a death certificate, try to rule out:
It will be fairly expensive, but I think it's a responsible use of tax dollars. It prevents family members from "wondering if it was their fault" or "deciding it must have been the immunization."
* Kids with febrile seizures are not at increased risk for SIDS, so it is probably unacceptable to invoke a seizure as causal: Arch. Dis. Child. 86: 125, 2002.
But after you've done all this, can your autopsy rule out smothering or all other forms of lethal trauma? In a word, "No" (Am. J. Dis. Child. 144: 137, 1990). The SIDS autopsy: J. Clin. Path. 45(S-11): 11, 1992.
* The pathology lab has nothing to offer for the differential diagnosis of "near-miss SIDS" (Am. J. Dis. Child. 140: 484, 1986). Protocol for the SIDS autopsy: J. Clin. Path. 40: 481, 1987.
When a second SIDS death takes place in a home, the pathologist's task is especially difficult. One big study concluded that most of these are natural, and that many are homicides, and that sorting them out definitively is impossible (Lancet 365: 29, 2005). Probably you have to call the child protection folks (Arch. Dis. Child. 88: 699, 2003).
* Unexpected infant deaths in Israel (no scene investigations, almost no autopsies; just cultures, bone surveys, and rarely labs for metabolic disease): Arch. Dis. Child. 92: 697, 2007.
* Unexpected infant deaths in England (full scene investigation, everybody gets autopsied, when autopsy results are back, team approach includes a sit-down talk with the family. Hurrah for England). Arch. Dis. Child. 95: 291, 2010.
CHRONIC OBSTRUCTIVE PULMONARY DISEASE (COPD, chronic air flow obstruction, * COAD, * CAWO, * chronic airway obstruction; review Lancet 379: ): The most common cause of activity-restricting disability in the U.S. Morphology and pathophysiology review explaining the obstruction: Lancet 364: 709, 2004. Clinical review (no surprises): Lancet 362: 1053, 2003.
"Chronic obstructive pulmonary disease" is a traditional and not-very-helpful grouping of four illnesses:
1. chronic bronchitis
2. emphysema
3. asthma
4. bronchiectasis.
The term "COPD" generates tremendous confusion, and I urge you not to use it. Most clinicians reserve it today for emphysema ("and chronic bronchitis"). We should probably remove bronchiectasis, and add obstructive and obliterative bronchiolitis.
"COPD" includes two very common "diseases" that typically occur together as a result of smoking.
CHRONIC BRONCHITIS causes obstruction to air flow because of edema, necrosis, fibrosis, and recurring infections in the bronchial tree.
The walls do get thicker and this narrows the bronchial lumens. This has been confirmed by some good pathology studies (Am. Rev. Resp. Dis. 143: 1152, 1991; Chest 97(S2): 6S, 1990), and now we see it on high-resolution imaging studies (it's the littlest airways, of course: AJRCCM 173: 1309, 2006). Since this is not a feature of the lungs of asymptomatic smokers (Radiology 235: 1055, 2005), it seems reasonable to think that this is the real lesion of a real disease.
Although narrowing of the bronchial lumens ("thickening of the bronchial walls") interferes some with breathing, it's usually the severity of the co-existing emphysema that really disables these people.
The vicious cycle of bacterial colonization, infection, and damage probably plays a part in the progression of the disease (AJRCCM 173: 991, 2006).
Increased mucous secretions and hyperplasia of mucous-secreting glands by themselves, though typical of chronic bronchitis, are now thought NOT to contribute significantly to obstruction (i.e., it's not prolonged choking to death on hockers). Am. Rev. Resp. Dis. 128: 491, 1983; Am. Rev. Resp. Dis. 133: 942, 1986.
EMPHYSEMA causes obstruction to air flow because loss of the lung's elastic recoil allows small airways to collapse at the beginning of expiration. We all lose some elasticity as we age, but smokers and people who are alpha-1 antitrypsin deficient lose it much more, and also have some destruction of their septa, which is not part of normal aging.
NOTE: Some people include all asthma cases as COPD, but regular asthma has a much better prognosis. Today, even chronic "asthmatic bronchitis" is distinguished from other forms of chronic bronchitis, which are much more lethal: NEJM 317: 1309, 1987.
Both emphysema and chronic bronchitis are most commonly caused
by CIGARET
Smokers should be prepared for: |
 I remember this one from the 1950's |
Robbins's past estimate of "10,000 to 20,000 deaths in the U.S." yearly due to COPD is much too low. There are about 2 million diagnosed emphysema patients in the U.S. at any time, and the real prevalence of this disease is much higher. The CDC lists 137,000 "chronic lower respiratory disease" USA deaths yearly (2009), mostly emphysema -- more than stroke (128,000) or accidents (118,000) or Alzheimer's (79,000).

EMPHYSEMA
|
|
|
|
|
|
|
|
|
Australian Pathology Museum High-tech gross photos
|
{11506} emphysema
{20239} emphysema
{20993} emphysema
{29153} emphysema
{38362} emphysema
{38365} emphysema
{49068} "pan-acinar emphysema"
{49069} "centrilobular emphysema"
The old-fashioned anatomists define "emphysema" as an abnormal, permanent dilatation of part of all of the acinus, with eventual destruction of many of the alveolar walls.
I predict that the definition will eventually be changed to reflect the true sine qua non -- loss of elasticity of the lung. The new definition may also include the requirement that there be some destruction of the alveoli.
You'll diagnose emphysema on lung function tests by noting prolongation of the time required for a full forced expiration, in the absence of asthma.
Two common forms are distinguished:
CENTRILOBULAR (centriacinar) EMPHYSEMA shows more dilatation of the respiratory bronchioles and their alveoli.
Traditionally, this is seen in early smoker's emphysema "because cigaret smoke exposure is heaviest in the centers of the acini". And "it is worse in the upper lobes, because they get more smoke exposure" (why?).
PANLOBULAR (panacinar) EMPHYSEMA involves the acinus uniformly.
Traditionally, this is caused by alpha1-protease inhibitor ("antitrypsin") deficiency "because the blood brings neutrophils to all of the lung uniformly". And "it is worse in the lower lobes, because they get more blood" (why?)
By the time emphysema patients come to autopsy, the damage is so severe that you won't be able to make the distinction, and it's not really something that matters to the clinician, radiologist, or patient.
The pathogenesis is pretty clear.
Emphysema is NOT caused by air trapping behind inflamed bronchioles (asthmatics don't develop emphysema from it) or playing wind instruments (Chest 88: 201, 1985).
The problem is damage to the elastic fibers of the lung by elastases from polys, monos, possibly pancreas. Of course, smoking cigarets brings lots of polys to the lung.
Deficiency of serum alpha-1-antitrypsin (our major * "serpin" -- serine protease inhibitor, gene SERPINA1) is an inborn error of metabolism ("serpinopathy") in which severe panacinar emphysema develops in non-smokers. Review NEJM 360: 2749, 2009; today's lab diagnosis Am. J. Clin. Path. 139: 184, 2013.
This results from inability to efficiently release an abnormal anti-protease from the hepatocytes. Inclusions develop in the cells. The defects is the result of alleles at the Pi ("protease-inhibitor") locus -- "M" is the common allele, while "S", "Z", and others result in less product getting into the bloodstream. Lab analysis Am. J. Clin. Path. 138: 398, 2012. Homozygous patients may also get cirrhosis. Whether heterozygotes are more at risk for emphysema if they smoke is the subject of much discussion, as they have intermediate levels of anti-protease. Replacement therapy with the enzyme ("Prolastin" or "Respitin") is now in use for these patients, at a cost of around $36,000 per year (Chest 119: 745, 2001; third-party payers are of course balking and estimates of the cost of a year of life saved vary widely: Chest 117: 875, 2000).
* Social factors in disease: The Swedes screened all newborns for SZ and ZZ and warned all parents of these children to stop smoking. They discovered that none of the parents complied (Thorax 43: 505, 1988).
 Huckleberry Finn smoking | Tobacco smoke augments elastases, inhibits anti-proteases, and promotes infection with further release of elastases. We do not know why some smokers get much worse emphysema than others. |
Because of loss of elastic recoil, small airways collapse during forced expiration.
The classic "emphysema" patient is a "pink puffer", with normal PaCO2, barrel chest, pursed lips, dyspneic, tachypneic, thin (he's working hard all the time), miserable. The only consistent finding on physical exam is slowing of forced expiration ("Can you blow out a match at six inches with your mouth wide open?").
Pink puffers learn to keep their lungs hyperinflated to keep the respiratory bronchioles from collapsing, and this eventually changes the shape of the chest itself ("barrel chest", "increased AP diameter", "increased total lung volume").
These people eventually start getting bacterial lung infections, and die of cor pulmonale, pneumothorax, or pneumonia; less often pulmonary thromboemboli.
At autopsy, the lungs are hyperinflated and relatively bloodless. Eventually, broken alveolar septa domate the picture, and many of the capillaries in the septa are gone (we don't really know why the latter occurs).
Under the microscope, you'll see a lot fewer septa than normal. Some is due to destruction of the septa, some is due to the overinflation that spreads them out. You'll very likely see many septa with their ends "floating free in the breeze"; if you have a good eye, their free ends may show a slight swelling (* confusingly called "clubbing").
Update on "what kills pink puffers / blue bloaters?" -- the Serbs do lots of autopsies, and found the usual lesions are cor pulmonale, pneumonia, and thromboemboli: Chest 136: 376, 2009. One dies of complications, not the disease itself.
In 1996, the NEJM (334: 1095, 1996) published an article showing that the newly-popular "lung reduction surgery" (lung-lift, like face lift or certain other cosmetic procedures to reverse sagging, $75,000) produces improved exercise ability for a while. Just as you'd expect from what you've learned about emphysema, people who have little inspiratory resistance (i.e., wide open bronchi and bronchioles) do nicely, while those with really badly-narrowed bronchi are not benefited (NEJM 338: 1181, 1998). Update NEJM 343: 239, 2000 (better exercise tolerance, no proof of longer survival). It is now very common and the benefits are obvious (Chest 123: 1838, 2003). Changing times: Medicaid decided every smoker on welfare did not have a "right" to this surgery, and surprisingly few people were outraged.
* No one knows quite what to make of this, but it now seems that the alveolar lining cells of the emphysema patient are much more likely to be senescent (i.e., relatively unable to continue dividing) than that of age-matched non-emphysema patients (more P16INK4a and p21CIP1/WAF1/Sdi1 -- Am. J. Resp. CCM 174: 886, 2006).
BULLOUS EMPHYSEMA produces air-filled blebs (if >2
cm, you can call them "bullae") containing little
or no lung tissue, usually at the apices, sometimes (but by no
means always) at the sites of old TB![]() scars.
scars.
Most cases probably result from common emphysema, with the inelastic lung "collapsing under its own weight" (Thorax 44: 533, 1989); the upper lobes have more contact more tobacco smoke because they are better ventilated.
The blebs may be removed surgically, with some relief of the "pink puffer"'s puffing.
Blebs are also prone to rupture, causing pneumothorax and sudden death. Iatrogenic disease: "IPPB breathing treatments" are irrational therapy for uncomplicated emphysema, and kill patients by blowing out blebs.
{10778} blebs in emphysema
![]() Emphysema patient
Emphysema patient
Barrel chest
Well-developed arms
Other forms of "emphysema":
* Paraseptal / distal acinar: rare, blamed for spontaneous pneumothorax in young people, especially folks with Marfan's and Marfanoinds. It causes marked expansion of single acini under the pleura with sparing of the others neary, and tends to follow the small interlobular septa nearby. I was skeptical for years about this but it's probably real: Am. J. Clin. Path. 136: 857, 2011.
Compensatory (i.e., following removal of a lobe of a lung, the other lobes expand; this is a misnomer, as there is no destruction of alveoli, and no loss of elasticity.)
"Irregular" or "tractional" (i.e., adjacent to contracted scar, etc.; another misnomer.)
"Senile" (loss of elasticity without loss of lung substance, from "old age")
"Interstitial emphysema" doesn't even refer to lung. It means air has been forced into the fibrous tissues of the body, often as the result of tearing of the lung itself (by real emphysema, by severe coughing, by a respirator, by a broken rib, by barotrauma). Listen for the "milkman's crunch" sign as the heart beats, palpate the little bubbles under the skin, and reassure the patient that it will reabsorb.
CHRONIC BRONCHITIS ("smoker's cough" -- never trivial)
Defined clinically, as persistent cough with sputum production for at least three months in at least two consecutive years. (Worth committing to memory.) Update Lancet 379: 1341, 2012.
It's not the cough
That carries you off,
It's the coffin
They carry you off in.
Again, the usual cause is cigarets. Marijuana isn't exactly good for the lungs either (gee whiz, JAMA 259: 966, 1988; NEJM 318: 547, 1988) but so far hasn't produced wards full of respiratory cripples.
The "classic chronic bronchitis patient" is a "blue bloater", with increased PaCO2, obese, edematous (cor pulmonale), cyanotic, producing much sputum, happy (CO2 narcosis.)
The distinguishing feature of this kind of patient is an acquired tolerance for the hypercarbia that poor ventilation (i.e., from emphysema) ultimately causes. Unlike "pink puffers" (who retain their hypercarbic drive), these patients no longer really struggle to breathe, so long as they have adequate oxygen.
Because of the poorer alveolar ventilation, (* and perhaps with a contribution from obstructive bronchiolitis scarring extending to involve the microvasculature), pulmonary hypertension supervenes earlier in "chronic bronchitis" patients than in emphysema patients. And especially in emphysema, the pulmonary hypertension is due primarily to destruction of the vessels, with few or no actual proliferative lesions (Chest 131: 874, 2007).
Exacerbations are common and are the usual reason for the person ending up in the hospital. Eighty percent of the time, this is due to a bacterial or viral infection. The deadliest are still probably S. pneumoniae and H. influenzae;
Death may also result from cor pulmonale or from apnea brought about by administration of supplemental oxygen (remember, hypercarbia no longer stimulates respiration in these patients.) A little dose of a sedative can stop their respiratory drive, so be careful.
Many clinicians treat chronic bronchitis with glucocorticoids "to control the component of bronchospasm, as in asthma". I suspect they're actually making the walls of the inflamed bronchi thinner, with even a little bit of widening the lumens helping a lot. As an autopsy pathologist in the 1980's, I was very impressed with the ability of systemic glucocorticoid side effects to kill these people. The pulmonologists finally switched to inhaled glucocorticoids at the end of the last century (Lancet 351: 773, 1998).
Genetic factors seem to determine whether a smoker becomes a "pink puffer" or a "blue bloater." See Am. Rev. Resp. Dis. 129: 207, 1984. The onset of "chronic bronchitis" is supposed to be earlier than emphysema, which is understandable since it will be diagnosed by hearing the patient rattle all those hockers, while emphysema is diagnosed only when shortness of breath becomes profound.
At autopsy we find copious secretions in the airways, even in the absence of pneumonia. The trachea itself may be filled with yellow slime.
Microscopically, we see thickening of the bronchial basement membrane (seen also in asthma), proliferation of goblet cells, hypertrophy and hyperplasia of mucous glands, more of the various inflammatory cells, more lymphoid aggregates and follicles than usual, and often a considerable amount of scarring (histopathology update on "smoker's small airways disease": NEJM 350: 2645, 2004). Even the cartilage may be thinned.
"Reid index" is ratio of thickness of submucosal mucous glands to entire submucosa. Normal is up to .4; increased in chronic bronchitis.
Only in the most severely crippled smokers, we see widespread obliteration of the lumens of the terminal bronchioles (i.e., obstructive bronchiolitis or denser scarring). See below.
{08761} chronic bronchitis (chronic inflammation, missing epithelium)
|
|
|
* "Coarse breath sounds" / "coarse rhonchi" are hockers moving around in the big airways. Make the patient cough, and they'll perhaps go away or at least change.
Despite elaborate systems of testing pulmonary function, the ultimate diagnosis of "COPD" is made on history and physical exam (Am. J. Med. 94: 188, 1993). This is true for most other diseases, too.
BRONCHIAL ASTHMA (Review for pathologists, emphasizing the pathology and the bewildering array of chemical and physiological abnormalities: Arch. Path. Lab. Med. 130: 447, 2006; update for science types Nat. Med. 18: 631, 2012 and several articles in this issue).
Common syndrome (10% of kids, 5% of adults; well-known to Hippocrates and a focus for Maimonides) in which the small bronchi are abnormally responsive to various stimuli that cause constriction and/or are considerably inflamed (usually both). This produces episodes of dyspnea, wheezing, cough.
You'll hear plenty of wheeze sounds through your stethoscope; the sound is air rushing through narrow airways, making noises like the wind section of the orchestra playing terribly out of tune.
Asthma kills around 3000 people in the U.S. each year. Very few dead adult asthmatics took good care of themselves (no-nonsense article: Thorax 44: 97, 1989); and asthma death is primarily an underclass phenomenon (NEJM 331: 1542, 1994). Likewise, the asthmatic children who die are for the most part the underclass ones, as a result of smoky homes and "disease mismanagement": Ped. Clin. N.A. 50: 65, 2003.
* There is a familial tendency and some known genes but this is of no clinical value (J. Allerg. 126: 200, 2010). One known locus is the interleukin 6 receptor (Lancet 376: 1014, 2011).
Mast cell factors appear to mediate the bronchoconstriction regardless of what triggers the attack.
These factors include histamine, bradykinin, leukotrienes ("SRS-A", etc.), prostaglandins (must be present: Science 287: 2013, 2000), probably others. We're still learning what's in those mast cells.
* If you actually look inside (rather than below) the epithelium, asthmatics reportedly average ten times as many intraepithelial mast cells as their non-asthmatic counterparts (Am. Rev. Resp. Dis. 148: 80, 1993).
All about arachidonic acid metabolites in lung disease: Am. Rev. Resp. Dis. 143: 188, 1991. Leukotrienes are of course notorious for being chemotactic for inflammatory cells, and for tightening smooth muscle.
Asthmatic attacks are often triggered by:
* "I wasn't surprised": Allergy shots offer no benefits for asthmatic kids getting appropriate medical treatment: NEJM 336: 324, 1997
* Omalizumab, the anti-IgE, helps hard-to-manage inner-city kids and young adults: NEJM 364: 1005, 2011.
ALLERGIC ASTHMA is said to be present when the patient's attacks are typically triggered by IgE-mediated hypersensitivity.
This often is severely disabling in childhood, though it generally gets better in adult life. An old misnomer is "extrinsic asthma".
Remember both the familiar inhalants and food allergy (J. Allerg. Clin. Imm. 112: 168, 2003).
Remember histamine, leukotrienes, prostaglandin D2, and platelet activating factor as the major contributors to this kind of wheezing, with leukotrienes perhaps most important.
Industrial asthma is a serious problem. The worst offenders are cedar wood platinum salts, anhydrides (epoxy hardeners) and isocyanates, followed by proteolytic enzymes, epoxy resins themselves, lab animals, vinyl chloride used in meat packing, flour, crab processing, oil mists, and penicillin. Formaldehyde asthma is a problem for a few unlucky pathologists. If you are asked to give an opinion as to whether a patient's asthma is due to their occupation, see the American College of Chest Physicians' standards at Chest 134(3S): 1S-41S, 2008.
* Platelet-activating factor antagonists: Chest 108: 529, 1995. Montelukast / zafirlukast for leukotriene receptor blockade (JAMA 279: 455 & 1181, 1998) -- works wonders for exercise-induced bronchoconstruction (chilling and drying?) and mild allergic asthma (NEJM 339: 1998).
We believe that some of the longstanding epithelial havoc is usually wrought by major basic protein of eosinophils recruited to the sites of the reaction. Be this as it may, another eosinophil protein crystallizes as "Charcot-Leyden crystals" in the sputum of allergic asthmatics.
If a chronic aspergillus![]() infection gets established in an
asthmatic's lungs,
allergy to this fungus is likely to make the
asthma much worse.
As many as 15% of severe
cases get this. In "allergic bronchopulmonary aspergillosis",
the fungi actually find safe haven inside the plugs, creating a vicious
cycle.
infection gets established in an
asthmatic's lungs,
allergy to this fungus is likely to make the
asthma much worse.
As many as 15% of severe
cases get this. In "allergic bronchopulmonary aspergillosis",
the fungi actually find safe haven inside the plugs, creating a vicious
cycle.
IDIOSYNCRATIC ASTHMA is said to be present when the patient's attacks are typically triggered by exposure to aspirin (Chest 88: 387, 1985), another cyclo-oxygenase inhibitor, and/or tartrazine yellow. These people's small airways are teeming with eosinophils and mast cells (Am. J. Resp. CCM. 153: 90, 1996, NEJM 346: 1699, 2002), the obvious source for the extra leukotrienes.
This is more likely to begin in adult life. An old misnomer is "intrinsic asthma". (Look for nasal polyps in the aspirin-sensitive patient.)
NOTE: There is a great deal of talk about asthma becoming more common than it was a few decades ago. You'll need to decide whether this is true, and if so, why.
NOTE: REACTIVE AIRWAYS DYSFUNCTION / DISEASE (RADS, acute irritant-induced asthma) is an important fairly-newly-recognized entity in which asthma and/or intractable cough follow a single noxious exposure of the bronchial mucosa to something hurtful (i.e. poison or hot gas, poisonous fumes).
The World Trade Center disaster helped show that this entity is very, very real (for example, NEJM 347: 806, 2002; Chest 125: 1284, 2004). Reactive airways following chlorine gas release: MMWR 61: 982, 2012.
* Truth be told, you'll find reading about the "pathogenesis of asthma" extremely frustrating because there's no one-size-fits-all account, and the immunopathogenesis seems completely different in different patients. See J. Allerg. Clin. Imm. 131: 1267, 2013. There's even a severe subtype rich in non-necrotizing granulomas (Am. J. Resp. CCM 186: 501, 2012).
Regardless of cause, the pathology in true asthma is inflammation of the bronchial mucosa, with eosinophils, and (probably also, maybe as a result) increased fragility of the epithelium. Review Chest 124: 32, 2003.
The smooth muscle is also likely to be hyperplastic. This (and some increase in collagenization) is the principal histologic feature correlating with severity (AJRCCM 167: 1360, 2003; comfirmed amazingly in Am. J. Resp. Crit. Care Med. 185: 1058, 2012).
Part of the trouble, we may reasonably think, is irritation of the C-fibers that travel up the vagus nerve and mediate bronchospasm by an autonomic reflex.
The worse the asthma, the worse the inflammation (Am. J. Resp. CCM. 154: 24, 1996). No surprise. Anatomy of asthma, with a focus on the long term "repair" changes that actually make things worse: J. Allerg. Clin. Imm. 98: S278, 1996.
Watch mepolizumab, an anti-interleukin-5 monoclonal antibody, for treating difficult asthma (NEJM 360: 973 & 985, 2009). Interleukin 5 is of course a major recruiter of eosinophils
Only a small percent of asthmatics lack eosinophils, and it's not clear whether this is the same disease: Am. J. Med. 115(S-A3): 49S, 2003; update J. Allerg. Clin. Imm. 119: 1043, 2007 (poorer response to steroids). In the animal model, the airways do not undergo much fibrous thickening if the animal has no eosinophils (Science 305: 1776, 2004).
Fatal cases ("status asthmaticus") show mucosal edema, many eos, polys, mucous secretions, desquamation, and obvious basement membrane thickening (see Thorax 87: 152-S, 1985).
A study of non-fatal asthma cases showed that all had thick bronchiolar epithelial basement membranes. Non-asthmatic controls had no mast cells or eosinophils, while these were present in most of the asthmatics. Asthmatics also showed more of a tendency for the epithelium to fragment, the cells dying and/or becoming detached. See Am. Rev. Resp. Dis. 145: 922, 1992.
The thickening of the basement membrane, hyperplasia of smooth muscle (seems T-cell-mediated Am. J. Resp. Crit. Care Med. 182: 317, 2010), increase in goblet cells, and hyperplasia of bronchial glands is now called "remodelling" and is the most-studied phenomenon in asthma (Am. J. Resp. CCM 164: S46 & S52, 2001; Chest 129: 1068, 2006; Am. J. Resp. CCM 176: 138, 2007; J. Allerg. 123: 1090, 2009; J. Allerg. 125: 1037, 2010); NEJM 364: 2058, 2011; the causes remain as mysterious as always, with many "immunologic and inflammatory mechanisms" all implicated (J. Allerg. Clin. Imm. 121: 560, 2008).
Sputum from most asthmatics with type I immune injury shows Charcot-Leyden crystals (eosinophil protein), Curschmann's spirals (coily strings of altered goo from little airways).
Chest deformities (barrel, pigeon, Harrison's sulcus) often result from severe childhood asthma (JAMA 259: 1722, 1988).
* Future pathologists: In patients with unexplained cough, the simple trauma of coughing can produce impressive airway mucosal inflammation even in the absence of diagnosable disease. Keep this in mind when reading biopsies: Chest 130: 362, 2006.
And just to make things more complicated, it turns out that one common cause of unexplained cough in adults with normal chest x-rays is NON-ASTHMATIC EOSINOPHILIC BRONCHITIS, with pathology similar to asthma but airways that are not hyper-responsive (Am. Fam. Phys. 84: 887, 2011). Puzzle THAT out. Pulmonologists will make the call on biopsy.
|
|
Australian Pathology Museum High-tech gross photos
|
{08228} asthma, hyperinflated lungs (notice
that they completely cover the heart in front)
{08231} asthma, mucus plugging airways
{08234} asthma, plugs
{29236} asthma, plugs
{07564} asthma, thick basement membrane
{08240} asthma, thick basement membrane, lots
of smooth muscle, lots of epithelial cells lying around
{08243} asthma, thick basement membrane,
mucus plug (trichrome stain)
{09899} Curschmann's spirals
{25999} Curschmann's spirals
{27492} occupational asthma; isocyanates
are infamous
The mainstay of treating asthma is inhaled glucocorticoids, which (when not abused) are incredibly safe (NEJM 332: 868, 1995; Thorax 49: 1185, 1995). Best used in low doses daily; they actually reverse the anatomic pathology (inflammatory cells, cytokine production, even basement membrane thickening) that allows acute attacks to occur (Am. J. Resp. CCM. 150: 17, 1994).
* Did you know that glucocorticoids cause eosinophils to undergo apoptosis? (Am. J. Resp. CCM. 154: 237, 1996)
* Acupuncture totally fails when tested as a treatment for asthma: Chest 121: 1396, 2002.
* Bronchial thermoplasty involves selective destruction of the smooth muscle by heat. It seems to help in hard cases (NEJM 356: 1327, 2007).
Common sense is making headways: London's physicians discovered that when hard-to-treat asthmatic children got their homes visited by a nurse, the problem was usually parents not complying (smoking, filthy homes, not giving the kids their medicine, need for "psychological referrals") -- and forcing the parents to change under threat of law prevented physicians from having to give stronger medicine (Arch. Dis. Child. 94: 780, 2009).
* Although I have not seen this in the literature, I
was told twice in the 1990's,
both times by members of "conservative religious family-values"
identity-groups, that children are only wheezing to get
attention and should be punished for it. If I'm hearing this,
then so are at least some of your patients.
I urge you to call this what it is -- one more stupid, ugly lie. I've
taught you how to deal with these things ("If
that were true, then wouldn't you be able to observe....?")
* Regular spinal manipulation is heavily promoted to the public by many chiropractors
for childhood asthma. Since childhood asthma almost always gets better by
itself, a controlled study would seem to be indicated --
and has been done (no measurable benefit: NEJM 339:
1013, 1998 -- in an issue with two positive studies of the
value of manipulation for low back pain.)
"All that wheezes is not asthma." Wheezing may also result from:
* This might be a good place to mention PEPPER SPRAY, used by law enforcement and private citizens. You may hear it alleged that this has killed people; I find this hard to believe and so do the more scientifically-minded reviewers (Am. J. For. Med. Path. 16: 1995).
OBSTRUCTIVE BRONCHIOLITIS ("constrictive bronchiolitis"; extreme cases are "obliterative bronchiolitis"; update NEJM 370: 1820, 2014)
Despite the similar name to "bronchiolitis obliterans" (now "cryptogenic organizing pneumonia"), this is a totally different entity, with dense fibrous tissue beneath the epithelium, rather than loose fibrous tissue within the lumens. Think about why the pulmonary function tests will give an obstructive pattern.
Look for underlying...
![]() Obliterative bronchiolitis
Obliterative bronchiolitis
Lung pathology series
Dr. Warnock's Collection
BRONCHIECTASIS ("ectasia", or "pulling wide" of the bronchi; Chest 134: 815, 2008; named by Laennec)
Defined to be the permanent cylindrical dilatation and ulceration of part of the bronchial tree.
The clinical picture is chronic cough with sputum production, up to many cups daily. (Some might put bronchiectasis is on a continuum with chronic bronchitis.)
The dilatation ("-ectasis") results from contraction of scar surrounding the bronchus, and from surrounding atelectasis.
|
|
|
|
|
{05762} bronchiectasis
{10787} bronchiectasis
{24542} bronchiectasis
{27242} bronchiectasis
{38383} bronchiectasis
{38389} bronchiectasis
Bronchiectasis complicates respiratory infections (severe, or those in the immunosuppressed -- remember pediatric AIDS), asthma, cigaret smoking, inherited disease (especially cystic fibrosis and primary ciliary dyskinesis/immotility, including Kartagener's.) If you aspirate a foreign body that stays in a bronchus, you'll probably end up with bronchiectasis in the region. Today, many cases are idiopathic.
In older days when bronchiectasis was much more common, it
often followed measles![]() ,
staphylococcal
,
staphylococcal![]() or pertussis pneumonia. Pertussis (look for lots of lymphocytes
if it comes to autopsy) is once again very much with us
thanks to the anti-immunization movement, and measles is enjoying
a resurgence in Europe (many thousands of cases yearly) and in Israel for the same reason.
The American Northwest has a terrible problem with pertussis MMWR 61: 517, 2012.
Thousands of kids are hospitalized in Denmark these days with pertussis; maybe 1 in 50
ends up with epilepsy from it (JAMA 314: 1844, 2015).
or pertussis pneumonia. Pertussis (look for lots of lymphocytes
if it comes to autopsy) is once again very much with us
thanks to the anti-immunization movement, and measles is enjoying
a resurgence in Europe (many thousands of cases yearly) and in Israel for the same reason.
The American Northwest has a terrible problem with pertussis MMWR 61: 517, 2012.
Thousands of kids are hospitalized in Denmark these days with pertussis; maybe 1 in 50
ends up with epilepsy from it (JAMA 314: 1844, 2015).
Regardless of the predisposing lesion, the proximate cause is a bacterial infection ("normal throat flora", H. influenzae, S. pneumoniae, anaerobes). Once established, bronchiectasis will persist due to a vicious cycle of exudation and infection.
Bronchiectasis results in serious social problems in many cases; it may lead to amyloidosis, brain abscesses, cor pulmonale, and so forth.
The anatomic pathologists sees bronchi pulled wide by contraction of scar surrounding the ulcerated airways. (Ignore the quaint subclassification of "saccular", "cylindroid", and "fusiform" bronchiectasis).
Modern medical and surgical treatment has improved the outlook for these patients greatly.
PRIMARY CILIARY DYSKINESIA SYNDROMES (Am. J. Resp. CCM. 151: 1559, 1995).
This is a huge topic that includes illnesses where the cilia have no dynein arms, and a few others. What they have in common is that the cilia do not beat properly. Genes for primary ciliary dyskinesia are of course mostly recessive: Am. J. Resp. CCM. 174: 128 & 858, 2006. Patients have:
When situs inversus is present we speak of "Kartagener's".
We will be glad to help you make the diagnosis by electron microscopy.
* The original Kartagener's transgenic mouse: Nature 354: 306, 1991.
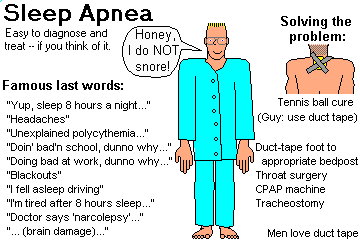
OBSTRUCTIVE SLEEP APNEA: NEJM 347: 498, 2002
Sleep apnea victims all snore famously (snoring correlates with excessive daytime sleepiness and other quality-of-life issues: Chest 110: 659, 1996). Most are overweight (more fatty tissue in the upper airway) or have huge tonsils or jaw deformities. Most sleep in the supine position. Most have "thick necks" and/or drink a lot (Am. Rev. Resp. Dis. 141: 1228, 1990; Thorax 46: 86, 1991). For some reason, premenopausal women are seldom affected.
* Worth remembering in treating patients: Obstructive sleep apnea often accounts for deterioration in patients with hypothyroidism (myxedema) and Down syndrome (Br. Med. J. 296: 1618, 1988).
As the victim starts to go into deep sleep, his upper airway closes (physiology: JAMA 266: 1384, 1991), he thrashes, snorts, partly wakes up, and re-opens the airway with a gasp. The cycle repeats every few minutes through the night, and the patient never gets to sleep soundly.
If you gasp or choke while you're asleep, you probably have sleep apnea. This correlates very closely with pricey sleep lab results: JAMA 310: 731, 2013.
Sleep apnea results in morning headaches, daytime somnolence, "narcolepsy", cognitive and behavioral changes, school and job failure, "psychiatric disease", divorce ("She refused to sleep with me any longer"), auto accidents (infamous, "I fell asleep with no warning" or "I blacked out": Arch. Int. Med. 151: 1451, 1991; NEJM 340: 847, 1999), "near-miss SIDS", sudden "cardiac" death, snooze angina (Lancet 345: 1085, 1995), hypertension (Br. Med. J. 320: 479, 2000; JAMA 307: 2169, 2012, lots more), etc.
* It is doubtful that sleep apnea takes many years off life expectancy, but it does have a very negative impact on quality of life (Chest 94: 531, 1988). Neglected, it exacerbates the common cardiovascular diseases (JAMA 290: 1906, 2003). Occasionally it does kill people (Chest 94: 9, 1988), and is now being "linked to dementia" (Chest 95: 279, 1989).
Most cases seem to involve subtle abnormalities of throat anatomy and mechanics: Lancet 337: 597, 1991 (* more type IIa muscle fibers in these people's throats), as well as problems with the reflexes that keep the throat open (Am. Rev. Resp. Dis. 143: 810, 1991).
* A higher-than-expected percentage also have lung damage from smoking, making the clinical picture more complex.
The hypoxemia, hypercarbia, and/or straining against a closed upper airway may produce deadly cardiac rhythm disturbances ("fatal heart attack while sleeping", maybe even "SIDS"). However, the large majority of people with sleep apnea do die while awake (Chest 94: 531, 1988).
For documentation, call the sleep lab and have them do polysomnography on your patient (* $1200).
Treatments:
Have the patient SLEEP ON ONE SIDE, so the uvula flops out of the way (South. Med. J. 79: 1061, 1986). If you like, duct-tape a tennis ball to the back of his neck.
The drug PROTRIPTYLINE helps the brain maintain a patent airway.
UVULOPALATOPHATYNGOPLASTY (an operation) and/or TONSILLECTOMY is moderately useful.
 | * Missionary physician-hero Dr. David Livingstone ("I presume?") cured himself of sleep apnea by cutting off his own uvula while in Africa. |
MACHINES that provide continuous positive airway pressure work wonders for most victims ($1000) -- Am. Rev. Resp. Dis. 137: 1238, 1988.
TRACHEOSTOMY is the last resort but cures sleep apnea. * Mini-tracheostomy for this disorder: Thorax 44: 224, 1989.
PICKWICKIAN SYNDROME ("obesity hypoventilation syndrome"): severe sleep apnea resulting in loss of hypercarbic drive (sleep-apnea's blue bloater). Patients are very obese and have high blood carbon dioxide levels even while wide-awake. I have wondered whether these patients have already suffered some brain damage from the hypoxic episodes, and hence overeat. * The name comes from Joe, the sleepy fat boy in Charles Dickens's "Pickwick Papers".
CENTRAL SLEEP APNEA is like obstructive sleep apnea, except that victims do not snore or struggle.
In others, the breathing centers aren't quite in synch, and patients Cheyne-Stoke.
In others (notably some folks with CHF), one might breathe super-hard after a brief rise in PaCO2, and stop breathing for a while after it's (over)corrected (NEJM 341: 949, 1999).
Patients complain of daytime somnolence, etc., but are easier to sleep with.
* Treatment with CPAP reportedly works well, though it's hard to explain why.
* We used to "call respiratory arrests" and do CPR on these patients ("saved lots of lives").
THE ONDINE'S CURSE or CENTRAL HYPOVENTILATION is a serious situation in which there is diminished respiratory drive from the brain. You'll learn about this in the ICU. There is a genetic form (* homeobox mutation, Nat. Genet. 33: 440 & 459, 2003) -- or a premature baby's respiratory centers may simply not be well-developed.
* By the way... CHEYNE-STOKES RESPIRATION -- so familiar in those who care for the dying and for those with congestive heart failure -- can be normal variant. If the person's healthy, do not confuse it with central sleep apnea or death.
Systemic high blood pressure and sleep apnea: Long-known, now well-demonstrated, but nobody knows why it happens. NEJM 342: 1378, 2000.
* I predicted in these notes in the 1980's that
"Gaisbock's primary idiopathic polycythemia" (long-noted to be
a disease of heavy, middle-aged
men) would prove to be due to sleep apnea. Anyway,
this diagnosis is almost never made any more, and sleep apnea is now
a well-recognized cause of polycythemia. So I think that's the explanation.
LUNG INFECTIONS
|
|
The airways of non-smokers are normally sterile below the vocal cords, but almost any organism capable of causing disease has caused PNEUMONIA, infection of the lung substance.
INFECTIONS BY THE COMMON BACTERIA most often cause exudation (edema fluid, then polys and maybe macrophages) into the alveolar spaces ("I'm coughing up icky stuff").
If the bacterial infection is confined to patches within individual lobes, it is called BROCHOPNEUMONIA.
If the bacteria spread aggressively ("through the pores of Kohn / canals of Lambert"), stopping only for interlobar fissures, it is called LOBAR PNEUMONIA.
|
|
|
|
|
|
|
|
|
|
|
|
VIRAL AND MYCOPLASMA INFECTIONS most often cause mild edema confined to the interstitium, and infiltration of the interstitium by lymphs and macrophages ("I've got a dry, hacking cough").
TUBERCULOSIS, PNEUMOCYSTIS, and FUNGAL PNEUMONIAS each have distinctive pathology.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
{11435} cryptococcal pneumonia![]()
|
|
The terminology for all this is a little confused. Often "pneumonia" is used for inflammation in the alveolar air spaces, and "pneumonitis" is used for inflammation limited to the interstitium.
Lung infections are a common pathway out of life for people with disabling diseases of all sorts.
For more on coccidioidomycosis, click here.
For more on cryptococcus![]() , click here.
, click here.
For more on histoplasmosis![]() , click here.
, click here.
BRONCHOPNEUMONIA ("lobular pneumonia")
This patchy lung infection is ubiquitous in the hospital, and you will spend much of your time diagnosing and treating "pneumonia".
Why sick people get bronchopneumonia:
The anatomic pathology is nodules of edematous to hemorrhagic-purulent, patchy infected areas in the lung substance. Often easy to see, they are always easier to feel, being distinctly firmer than normal lung. You can even hear that there is less air.
Microscopically, the lesion is polys and fibrin nets in the air spaces. There is seldom much fibrin. (Important exception: E. coli pneumonias are mostly interstitial.)
Bronchopneumonia used to be a common complication of measles![]() and whooping cough.
Secondary bronchopneumonia caused by staph
and whooping cough.
Secondary bronchopneumonia caused by staph![]() was a major killer in the 1918
influenza
was a major killer in the 1918
influenza![]() pandemic.
pandemic.
PNEUMONIC PLAGUE
develops during plague![]() outbreaks,
and is transmitted person-to-person without requiring a rat flea.
The virulence factor turns out to be plasminogen activator (think
about it: Science 315: 529, 2007).
outbreaks,
and is transmitted person-to-person without requiring a rat flea.
The virulence factor turns out to be plasminogen activator (think
about it: Science 315: 529, 2007).
|
|
|
|
|
{10148} bronchopneumonia (patchy)
{49076} bronchopneumonia
Though common, bronchopneumonia is an opportunistic infection, and patients are already sick with something that allows bacteria to gain a foothold.
Many people with bronchopneumonia have deficient gag and/or cough reflexes, from old age, anesthesia, drugs, pain, wasting diseases, paralysis.
If the cilia are not functioning optimally (hereditary dyskinesis, mild squamous metaplasia, cigaret smoking, irritant gas exposure, viral chest cold), it is hard to clear bacteria.
Alcohol and tobacco are noted for interfering with the ability of the alveolar macrophages to kill bacteria, and even oxygen therapy is supposed to be able to do this.
When secretions pool in the chest (bad chronic bronchitis, cystic fibrosis, behind an obstructing cancer, or just not being able to cough very well), bacteria have a great place to grow.
Edema fluid is a good culture medium, and it is common for a single clinical episode of bronchopneumonia to be series of infections by different organisms. It may begin as a pneumococcal infection, which is replaced by a H. 'flu infection, which is replaced by a klebsiella infection, which is replaced by a serratia infection. This probably explains most "antibiotic failures".
ASPIRATION PNEUMONIA / ASPIRATION PNEUMONITIS (NEJM 344: 665, 2001) result from inability to protect the airway from oral bacteria and stomach acid respectively. Newborns can aspirate meconium. All can be very severe.
|
|
|
|
|
Actually most bronchopneumonia cases are caused by bugs aspirated from the mouth.
The large majority of aspiration pneumonias are on the right side, and most often the right upper lobe is involved (why?).
* Fears that the "back to sleep" campaign for SIDS prevention would result in an increase in deaths from aspiration have proved unfounded (Pediatrics 109: 661, 2002).
* If you perform pulmonary lavage on a child with lung disease, you may be given the "lipid-laden macrophage index". This has been promoted recently as a marker for chronic aspiration; I am more willing to believe that it's less specific and just as likely to be the result of some alveoli being chronically obstructed (Eur. Resp. J. 18: 790, 2001).
Special cases:
{12638} vicious case of staph![]() pneumonia
pneumonia
![]() Pneumonia
Pneumonia
never mind the exotic bug...
Pittsburgh Illustrated Case
LOBAR PNEUMONIA
As noted above, this is an infection of an entire lobe produced by a virulent micro-organism.
By far the most common etiologic agent has always been
STREPTOCOCCUS PNEUMONIAE![]() ("pneumococcus", a gram-positive diplococcus called "captain of
the men of death", "the old man's
friend", etc.), which struck down healthy people in their prime
(Chest 99: 2, 1991). But this
organism is still easy to handle with antibiotics,
epidemics have become
rare, there's the vaccine, and few people die today of
pneumococcal pneumonia in the developed nations.
Worldwide, of course, it's still a major killer: Lancet 374: 1543, 2009.
("pneumococcus", a gram-positive diplococcus called "captain of
the men of death", "the old man's
friend", etc.), which struck down healthy people in their prime
(Chest 99: 2, 1991). But this
organism is still easy to handle with antibiotics,
epidemics have become
rare, there's the vaccine, and few people die today of
pneumococcal pneumonia in the developed nations.
Worldwide, of course, it's still a major killer: Lancet 374: 1543, 2009.
{27689} pneumococci in sputum; gram stain
|
|
|
KLEBSIELLA PNEUMONIAE ("Friedlander's pneumonia", named by German pathologist Carl Friedlander whose relationship if any to your lecturer remains unknown) causes lobar pneumonia in deteriorated alcoholics. This gram-negative rod is coated by a thick slimy capsule, and victims cough up sticky slime ("cranberry sauce").
|
|
Staphylococcus![]() (after influenza
(after influenza![]() ), H. 'flu,
pseudomonas, and
others are notable causes of lobar
pneumonia in those with damaged lung defenses.
), H. 'flu,
pseudomonas, and
others are notable causes of lobar
pneumonia in those with damaged lung defenses.
![]() Pseudomonas pneumonia
Pseudomonas pneumonia
Yutaka Tsutsumi MD
In the era when huge numbers of people died of pneumococcal pneumonia, the classical anatomic pathologists distinguished four successive stages of lobar pneumonia:
1. HYPEREMIA AND EDEMA: the bugs divide like crazy, and the blood vessels dilate and leak fluid in response to injury. "Congestion", given in traditional accounts of pneumonia, is an obvious misnomer.
2. RED HEPATIZATION: the inflammation progresses, and the damaged vessels now leak fibrinogen, which forms fibrin meshworks in the alveoli. Some red cells are released by damage to the blood vessels, and polys come in to fight the bacteria. The alveolar exudate becomes "rusty sputum".
3. GRAY HEPATIZATION: fibrin dominates the picture, while polys and red cells break down ("gray" because hemorrhage is no longer taking place and the red cells have lysed). Fibrin may ball-up in the alveoli as "fibrin knots".
4. RESOLUTION: plasmin clears out the fibrin, and the lung returns to normal (hopefully).
COMPLICATION: The pleural surfaces overlying the infection are almost always involved, accounting for the pain of lobar pneumonia. There will be fibrinous adhesions, which may resolve or turn into scars.
COMPLICATION: In Klebsiella, staph![]() ,
or pseudomonas
lobar pneumonia, there is often necrosis
and abscess formation, which greatly complicates healing.
Necrosis is rare in pneumococcal
pneumonia (exception: the slimy type 3). More on this below.
,
or pseudomonas
lobar pneumonia, there is often necrosis
and abscess formation, which greatly complicates healing.
Necrosis is rare in pneumococcal
pneumonia (exception: the slimy type 3). More on this below.
|
|
![]() Lung Abscess
Lung Abscess
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
{12641} staph![]() producing abscess
producing abscess
COMPLICATION: If the infection gets really bad in the pleural space, it will fill with pus ("empyema") and this will need to be drained. (This was the most helpful thing that a doctor could do for pneumococcal pneumonia before penicillin.)
![]() Suppurative pleuritis
Suppurative pleuritis
Not quite empyema
WebPath
{49077} empyema
COMPLICATION: Sometimes the fibrin in the alveoli mostly organizes instead, leaving a scar.
{39530} organizing pneumonia; balls of fibrin in the alveoli
COMPLICATION: The bacteria often spread to other structures (pericardial sac, meninges)
Today, uncomplicated lobar pneumonias are easily treated with antibiotics once the etiologic agent is identified.
{11431} lobar pneumonia
{11732} lobar pneumonia
{11733} lobar pneumonia
{11734} streptococci in lobar pneumonia,
gram stain
{12464} lobar pneumonia
{17565} lobar pneumonia
{17567} lobar pneumonia
LEGIONNAIRE'S DISEASE (legionellosis, "Pontiac fever", etc.)
This is a special form of bronchopneumonia named for a lethal outbreak at an American Legion convention at a hotel in Philadelphia.
The etiologic agent is LEGIONALLA PNEUMOPHILA. You need special stains (immunostain or silver) to see it.
The organism is common in standing water, especially in air-conditioning systems.
Most healthy people merely experience a bad "chest cold", but in older people who drink and smoke heavily, it is likely to be fatal if untreated.
* Legionellosis has reappeared as one of the opportunists in people treated with anti-TNF drugs (Am. J. Inf. Control 40: 470, 2012).
{08179} legionella demonstrated with a silver stain
|
|
NOTE: We are now recognizing that many cases of tough-to-treat
community acquired pneumonias
are due to CHLAMYDIA![]() (Arch. Int. Med. 148: 1425, 1988),
notably the TWAR strain
(CHLAMYDIA PNEUMONIAE, Chest 95: 664, 1989; NEJM
323: 1546, 1990). These infections have
long been recognized in newborns, who acquire the more familiar
sexually-transmitted pathogen
from the birth canal. It's now a notorious, chronic, hard-to-eradicate
infection of the airways (J. Inf. Dis. 182: 1678, 2000; Am. J. Resp.
CCM 164: 536, 2001).
(Arch. Int. Med. 148: 1425, 1988),
notably the TWAR strain
(CHLAMYDIA PNEUMONIAE, Chest 95: 664, 1989; NEJM
323: 1546, 1990). These infections have
long been recognized in newborns, who acquire the more familiar
sexually-transmitted pathogen
from the birth canal. It's now a notorious, chronic, hard-to-eradicate
infection of the airways (J. Inf. Dis. 182: 1678, 2000; Am. J. Resp.
CCM 164: 536, 2001).
* NOTE: Untreated syphilis![]() in the mother can cause necrosis of the lungs
of the unborn child (Arch. Path. Lab. Med. 126: 484, 2002.
in the mother can cause necrosis of the lungs
of the unborn child (Arch. Path. Lab. Med. 126: 484, 2002.
PNEUMOCYSTIS PNEUMONIA
This is a curious lung infection caused by a unicellular organism (somewhere on the lineage that split to form protozoa and fungi), PNEUMOCYSTIS JIROVECII (CARINII).
It was originally identified as the cause of plasma-cell pneumonia in malnourished children in Europe at the end of World War II.
It is now familiar as a cause of pneumonia in AIDS patients and people on cancer chemotherapy.
Instead of a plasma-cell pneumonia, these people show no
visible cellular reaction to the infection.
The organism and its cysts grow in the foamy exudate.
* As junior medical students,
we used to go to the airport to
pick up the pentamidine the CDC
shipped in every time we suspected pneumocystis. It was the only
thing that we knew worked.
If you spend any time on the oncology service, you will probably meet pneumocystis (i.e., you will develop antibodies), but it can't hurt you as long as your T-cells are working properly.
Future pathologists: You still need silver stains or immunostains if you want to identify pneumocystis with confidence.
{00456} pneumocystis pneumonia
|
|
|
|
LUNG ABSCESS
Polys plus necrosis in a confined space (i.e., walled off by granulation tissue) within the lung. Mechanisms (after Baby Robbins):
1. Aspiration of bacteria (bad teeth, tonsils), as when drunk or unconscious.
2. Complication of necrotizing pneumonia
(staph![]() ,
klebsiella,
pseudomonas, legionella) or
bronchiectasis
,
klebsiella,
pseudomonas, legionella) or
bronchiectasis
3. Obstructed bronchus (as, behind a cancer)
4. Infection within a lung cancer itself
5. Septic pulmonary embolus (leg vein infection, endocarditis, and now an extremely common problem of IV drug abusers -- Chest 94: 251, 1988)
6. Infarction of a pre-existing infection (as when a pulmonary embolus hits an area of bronchopneumonia)
Anaerobic bacteria are often present, and aerobes may also be involved.
Sooner or later, the enlarging abscess ruptures into an airway. The patient gets a worse cough and bad breath, while the radiologist looks for air-fluid levels.
VIRAL AND MYCOPLASMAL PNEUMONIA ("primary atypical pneumonia", "chest cold", etc.)
A family of infections by micro-organisms smaller than familiar bacteria, all causing interstitial pneumonitis.
A while back, a group biopsied a bunch of folks with colds, and found what you'd expect -- inflammation, mostly lymphocytes, in the bronchial mucosa (Am. J. Resp. Crit. Care Med. 151: 879, 1995).
![]() Viral lung infection
Viral lung infection
Lymphocytes
WebPath Photo
Many annoying chest-colds are caused by MYCOPLASMA PNEUMONIAE (check blood for cold agglutinins).
Viral infection of the lungs can be seen in measles![]() (major
killer worldwide), influenza
(major
killer worldwide), influenza![]() (all strains),
chicken pox, metapnemovirus & adenovirus
(all strains),
chicken pox, metapnemovirus & adenovirus![]() infection (look for "smudge cells"
with masses of virus making the nucleus look homogeneous; they can cause
serious pneumonia even in healthy people, Am. J. Med. Sci. 325:
285, 2003).
infection (look for "smudge cells"
with masses of virus making the nucleus look homogeneous; they can cause
serious pneumonia even in healthy people, Am. J. Med. Sci. 325:
285, 2003).
An especially vicious case of influenza will result in necrosis of all the bronchial epithelium, inviting staph superinfection. All in a large series of US H1N1 deaths autopsied had diffuse alveolar damage (ARDS): Am. J. Path. 177: 166, 2010; similar series Am. J. Clin. Path. 134: 27, 2010). The variety of other necrotizing lesions of the airway epithelium is described in Am. J. Resp. Crit. Care Med. 181: 72, 2010.
Something that's getting more recognition lately is that herpes simplex I is prone to erupt as a very serious pneumonitis in patients on long-term mechanical ventilation. Consider this the ultimate "stress lip blister." See Am. J. Resp. Crit. Care Med. 175: 935, 2007.
Adenovirus serotype 14 (Ad-14) is a putative emerging pathogen, an unusually severe viral pneumonitis. There have been outbreaks in military boot camps, but it does not seem to be overly serious in otherwise-healthy people (J. Inf. Dis. 203: 1388, 2011).
Viral pneumonia review: Lancet 377: 1264, 2011. To this day, no one knows how to tell a bad viral pneumonia from a bacterial pneumonia before autopsy, or whether to throw antibiotics at obvious chest colds.
For more on adenovirus, click here.
For more on influenza, click here.
For more on measles, click here.
For more on respiratory syncytial virus, click here.
|
|
|
|
|
|
|
|
|
|
|
RESPIRATORY SYNCYTIAL VIRUS![]() , once considered merely the usual
cause of "bronchiolitis" in toddlers,
is now known to be prevalent and
lethal among older adults.
, once considered merely the usual
cause of "bronchiolitis" in toddlers,
is now known to be prevalent and
lethal among older adults.
RSV remains a deadly illness in the poor nations, and is very common among children (Kenya -- JAMA 303: 2051, 2010).
You should already be familiar with the multinucleated epithelial cells in the bronchioles. You may see intracytoplasmic inclusions bodies.
* Interestingly, a vaccine was tried in the late 1960's and proved to make the disease worse instead of rendering the kids immune (J. Inf. Dis. 167: 553, 1993). It's finally clear how it happened: Nat. Med. 15: 21 & 34, 2009. The useless antibodies elicited by the vaccine covered and protected the virus, and generated immune complexes in the process.
* Parainfluenza pneumonias that prove fatal (i.e., the person is immunocompromised) may show syncytial giant cells as in RSV.
METAPNEUMOVIRUS, a cause of wheezing mostly in youngsters, was discovered in 2001 (Lancet 360: 1393, 2002; NEJM 350: 443 & 451, 2004; NEJM 368: 633, 2013). It's now in a tie with RSV as the most common viral infection in the transplanted lung (AJRCCM 178: 876, 2008). Like influenza, it's more of a winter virus; people get the infection again and again. The anatomic pathology is like other virus pneumonitis pictures, and you can see smudge cells as in adenovirus infection.
Lethal INFLUENZA![]() infection without staph superinfection presents
primarily as necrosis along the epithelium of the bronchi
and bronchioles. There may also be diffuse alveolar damage.
If the patient comes to autopsy, you will make the diagnosis by immunohistochemistry (Clin. Inf. Dis. 43: 132, 2006).
Unlike many other viral infections, there is no "trademark" histopathologic lesion.
infection without staph superinfection presents
primarily as necrosis along the epithelium of the bronchi
and bronchioles. There may also be diffuse alveolar damage.
If the patient comes to autopsy, you will make the diagnosis by immunohistochemistry (Clin. Inf. Dis. 43: 132, 2006).
Unlike many other viral infections, there is no "trademark" histopathologic lesion.
The severe H5N1 strain owes its deadliness, at least in part, to "cytokine storm" affecting the lungs. In the mouse model, PGE2 inhibition protects against this, and it is likely this will find clinical use (NEJM 359: 1621, 2008). Autopsy histopathology series: Am. J. Clin. Path. 133: 380, 2010.
HERPES SIMPLEX![]() can present as an ulcerative tracheobronchitis
in moderately immunocompromised hosts. Alternatively,
herpes simplex and herpes zoster
can present as an ulcerative tracheobronchitis
in moderately immunocompromised hosts. Alternatively,
herpes simplex and herpes zoster![]() can
both present as miliary hemorrhagic areas in the lung parenchyma
if immunosuppression is severe. Look for "herpes cells" with
a single intranuclear inclusion
can
both present as miliary hemorrhagic areas in the lung parenchyma
if immunosuppression is severe. Look for "herpes cells" with
a single intranuclear inclusion![]() surrounded by a clear halo.
(* In contrast to skin and cervix, these viruses seldom
produce multinucleation in the lung.)
surrounded by a clear halo.
(* In contrast to skin and cervix, these viruses seldom
produce multinucleation in the lung.)
The nastiest infectious pneumonitis in the U.S. is the HANTAVIRUS, from inhaled mouse droppings (originally though inaccurately "Navajo pneumonia"; we prefer "hantavirus pulmonary syndrome"; anatomic pathology Am. J. Path. 146: 552, 1995; review NEJM 330: 949, 1994). The viruses are found throughout the USA but most commonly causes illness in the southwestern states (the Sin Nombre virus variant infects the western deer mouse). These hantaviruses specifically damage the endothelial cells, producing a rapid-onset, extreme pulmonary edema. Distinctive for this infection is an abundance of immunoblasts in lung and peripheral blood. We await effective treatment, and perhaps 1/3 of diagnosed cases are fatal. There was an outbreak in Yosemite Park in September 2012.
SARS is a coronavirus: NEJM 348: 1977 & 1995, 2003. The origin remains unknown; the original claim that it was a zoonosis from eating wild animals didn't work out (Science 301: 1031, 2003). The epidemic: Lancet 362: 1353, 2003. Thankfully only about 1000 people died (follow-up Nat. Med. 9(s): S-88, 2004). The pathology (Lancet 361: 1773, 2003; Am. J. Clin. Path. 121: 574, 2004; Hum. Path. 36: 303, 2005; Am. J. Path. 170: 1136, 2007) features:
![]() SARS -- ARDS and giant cell
SARS -- ARDS and giant cell
CDC
Wikimedia Commons
* The receptor for the virus in the lung is angiotensin converting enzyme-2 (Nat. Med. 11: 875, 2005).
Middle-east respiratory syndrome coronavirus (MERS-CoV): NEJM 368: 2487, 2013; Lancet 381: 2265, 2013. The pneumonia may be severe, and kidney failure is also a feature. There are no human pathology reports yet (Sept 2013); the rhesus monkey gets an interstitial pneumonitis the virus reproducing primarily in the alveolar pneumocytes (PNAS August 2013). Curiously, despite the fear that it's generated, only about 5% of household contacts seem to catch even a subclinical illness: NEJM 371: 828, 2014, but transmission in the health care setting was common NEJM 372: 846, 2015.
* Nobody knows how many nasty chest-colds are really zoonotic
Q-FEVER![]() (Coxiella). Probably underdiagnosed. Pathologists look for "ring / donut granulomas".
"Poker player's pneumonia", the scourge of one town,
was Q-fever traced to a cat's placenta (NEJM
319: 354, 1988).
(Coxiella). Probably underdiagnosed. Pathologists look for "ring / donut granulomas".
"Poker player's pneumonia", the scourge of one town,
was Q-fever traced to a cat's placenta (NEJM
319: 354, 1988).
In most cases of viral pneumonitis that come to autopsy, the pathologist sees edema and inflammatory cells, and the process is confined to the interstitium.
The lungs are heavy but not airless (why not?) The inflammatory infiltrate is mostly lymphs and macrophages.
In severe cases, an ARDS picture supervenes, with hyaline membranes.
* LYMPHOID INTERSTITIAL PNEUMONITIS is a dense, polyclonal ("pseudolymphoma") infiltration of lymphocytes strictly confined to the pulmonary interstitium, usually in kids with AIDS or adults with Sjogren's. Don't confuse it with a virus. Some patients have a low-grade clonal lymphomas, which look identical; perhaps it's a spectrum (Chest 122: 2150, 2002).
|
|
|
![]() Viral Pneumonia
Viral Pneumonia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
{38402} bad viral pneumonia; the lung is nearly solid from all the inflammatory stuff in the alveolar septa
|
|
Most patients recover without treatment. The most worrisome thing about chest viruses for healthy adults is that they predispose the lung to bacterial superinfection.
You're already familiar with Kaposi's virus infection ("Kaposi's sarcoma") of the lung (Radiology 195: 545, 1995).
![]() Kaposi's in the lung
Kaposi's in the lung
Lung pathology series
Dr. Warnock's Collection
TUBERCULOSIS![]() ("TB", "the white plague" -- covered twice in
R&F). Update Lancet 378: 57, 2011. Also here.
("TB", "the white plague" -- covered twice in
R&F). Update Lancet 378: 57, 2011. Also here.
|
|
|
|
|
|
|
The idea that the body over-responds to the hated organism has been reconfirmed by the discovery that genetically-modified non-virulent mycobacteria still infect, but do not recruit other cells, and thus cause little harm (NEJM 360: 2471, 2009).
PRIMARY TUBERCULOSIS occurs when the TB bacillus first infects a person.
A single lesion (the GHON FOCUS) occurs just under the pleura in the midportion (midway between apex and base -- the best-ventilated area) of one lung.
![]() Tuberculosis
Tuberculosis
Very large Ghon focus
WebPath Photo
The bacilli find their way to the regional lymph nodes, and in a few weeks, granulomas have walled off the bacilli in both locations. (The combination of lesions in the lung and node is called the GHON COMPLEX). Viable bacilli remain in the Ghon focus/complex for life.
{08333} TB, lymph node
{08336} TB, lymph node
Much primary TB is asymptomatic, i.e., you discover you've turned your TB skin test.
PROGRESSIVE PRIMARY TUBERCULOSIS is the name given to overwhelming primary infection, which is not so rare as we used to teach.
The classic dogma is that almost all progressive adult TB represents reactivation of a latent primary focus. I have never understood why people believed this, and it turns out that it's clearly not true: NEJM 330: 1697, 1703 & 1710, 1994 (at least in America's slums; confirmed NEJM 346: 1453, 2002; TB in immigrants does often seem to be reactivation). Only in 2002 did we get molecular proof that one patient's TB had reactivated: J. Inf. Dis. 185: 401, 2002. Primary or re-activated, the pathology of bad TB is that of classic "secondary tuberculosis"....
SECONDARY TUBERCULOSIS ("active TB", "postprimary tuberculosis", "adult tuberculosis", "reinfection tuberculosis", "cavitary tuberculosis") occurs when bacilli escape the original Ghon focus or more bacilli enter the body from outside.
{08459} cavitary TB
{10230} TB in the lung, good cavity
{21151} disseminated, miliary TB in liver
|
|
The bacilli may be released from the Ghon focus by invading cancer or "by immunosuppression" of some sort.
The infection usually reappears at the apex of one or both lungs ("Simon's foci" -- the TB bacilli
actually entered the bloodstream, but grow best in the lung apex where oxygen is most
abundant.) This is one cause of "white pneumonia" ("pneumonia alba"; the other is
syphilis![]() ).
).
The better the patient's cell-mediated immunity, the more classic the granulomas.
In other words, the worst TB cases in America today have very few, if any, granulomas! For example, an AIDS patient can have lungs teeming with "red snappers" with only a sprinkling of macrophages trying to fight them. (This patient may not even be very sick, but his ability to transmit the infection is impressive.)
Polys are most abundant when the caseum has eroded into the large airways (common bacteria can enter the dead material). Also remember that TB tends to calcify (handy for radiologists who want to tell it from cancer).
{05949} TB eroding through the chest wall
{08187} TB, kidney
{08188} TB, lung
{08190} TB, lung
{08193} TB, lung
ARRESTED TB is secondary TB that has "calcified" and/or been largely replaced by collagen.
{11423} old TB
By contrast, PROGRESSIVE PULMONARY TUBERCULOSIS spreads throughout the lungs and can produce a "tuberculous empyema" by involving the pleural cavities.
When a large portion of the lung has undergone caseous necrosis![]() ,
extension into a large airway
causes all the debris to be coughed up -- exactly what the TB bacillus "wants", since this is how it is
transmitted to other people. The result is a CAVITY. (* Howler in R&F: "coin lesions" are lone
granulomas, not cavities.)
,
extension into a large airway
causes all the debris to be coughed up -- exactly what the TB bacillus "wants", since this is how it is
transmitted to other people. The result is a CAVITY. (* Howler in R&F: "coin lesions" are lone
granulomas, not cavities.)
TB is also prone to infect the larynx and larger airways ("tracheobronchial TB") and, because bacilli are swallowed, to affect the intestine (* look for Peyer's patches with ulcers having their long axes perpendicular to the long axis of the bowel).
MILIARY TB results when many TB bacilli enter the bloodstream but the granulomatous response is good. It is supposedly named for millet seed (the world fourth important grain crop -- * I've wondered of the origin could be "milli-", meaning "thousands" of little granulomas?).
When TB ruptures into a pulmonary artery, there is miliary involvement of part of a lung, while rupture into a vein results in miliary involvement of the rest of the body.
|
When I have fears that I may cease to be
|
Keats went to medical school for a year and was licensed as a physician and surgeon. He quit to devote his life to writing poetry. In this sonnet, he shares his fear that he may die before had has time to get all his ideas into writing. With his medical training, he had good reason to be afraid. A few days before he wrote the poem, he had experienced his first episode of hemoptysis. Three years later he was dead of tuberculosis. |
The very fact that we can talk about "TB and the arts" in the developed world is in stark contrast with the reality of several million deaths from TB alone or TB/HIV, especially in Africa (J. Inf. Dis. 205 S2: S340, 2012).
TB continues rampant among America's underclass and new-immigrant communities. The return of the mobile clinic and door-to-door screeners: Am. J. Pub. Health 103: 1292, 2013.
Actually making the diagnosis in questionable cases is difficult even in the era of molecular medicine. It takes several weeks to grow the microbe. Today's DNA techniques aren't very sensitive; gene studies to detect resistance are pretty good for rifampin but not for isoniazid (Arch. Path. Lab. Med. 138: 812, 2013).
The principal parasitic disease of the lungs is paragonimiasis, a mimic for TB that is most common in the Far East but is also a major problem in parts of Africa and South America. It is contracted from eating poorly-cooked crustaceans. It's easily mistaken for TB, especially when the eggs cause necrosis to get caughed up leaving a cavity. There are labs and especially seeing eggs in the sputum clinches the diagnosis. There are thirty or so species, one of which you can meet in Missouri (P. kellicotti; Am. J. Trop. Med. 84: 1005, 2011).
Reminder: In most chronic inflammatory diseases of lung, clubbing of the nails ("Hippocratic change") often develops because megakaryocytes embolize through the new vascular channels formed in the lungs.
INTERSTITIAL RESTRICTIVE LUNG DISEASE ("stiff lung"; "fibrosing alveolitis"; "honeycomb lung")
|
|
A generic term for damage leading to fibrosis of the alveolar walls (reviews for pathologists Arch. Path. Lab. Med. 135: 780, 2011; Arch. Path. Lab. Med. 136: 1234, 2012). Pulmonary compliance decreases, ventilation and perfusion are mismatched (i.e., blood flows through unventilated scar tissue), a mechanical barrier to oxygen exchange comes into existence (classically called the "diffusion barrier", "alveolar capillary block" -- in the past it has been over-emphasized but we do know it is a problem during exercise), and pulmonary blood pressure goes up (leading to cor pulmonale and death).
Fibrosis finally wipes out groups of alveoli. In most of these entities, the process is uneven throughout the pulmonary parenchyma, with some less-involved airways (especially respiratory bronchioles) stretched wide by scar contraction -- hence the radiographic and autopsy diagnosis of "honeycomb lung".
Regardless of cause, most fibrosing lung diseases are worst in the lower lobes, where there is ordinarily less air and more vasculature. (Asbestosis, a pneumoconiosis, can be an exception -- why?)
![]() Honeycomb Lung
Honeycomb Lung
Great gross photos
Dr. Warnock's Collection
Clinicians hear distinctive dry "velcro crackles" in pulmonary fibrosis.
The known etiologies of diffuse pulmonary fibrosis:
RHEUMATOID LUNG, SCLERODERMA LUNG, SJOGREN'S LUNG, ETC. ("connective tissue disease-associated lung") : secondary to autoimmune disease.
* If you are trying to tell a "secondary UIP" lung from idiopathic pulmonary fibrosis (which is even more ominous), and you can't decide based on the history, here's some helpful observations (Arch. Path. Lab. Med. 136: 1253, 2012):
 Rheumatoid lung, capillaritis
Rheumatoid lung, capillaritis
Lung pathology series
Dr. Warnock's Collection
ASBESTOSIS, BERYLLIOSIS, HARD METAL PNEUMOCONIOSIS (the latter is a hard carbide containing nickel and cobalt, famously used in cutting diamonds and elsewhere in the tool-and-die industry), and rare cases of longstanding FARMER'S LUNG ("hypersensitivity pneumonitis": T-cell havoc Chest 104: 38, 1993): due to pneumoconiosis
HISTIOCYTOSIS X ("Langerhans' cell histiocytosis" is a better name for this family)
DESQUAMATIVE INTERSTITIAL PNEUMONITIS: ("DIP") in addition to fibrosis, the alveoli clog with lipid- and mucin-laden macrophages. Most patients are smokers. This is now clearly a different disease from UIP / idiopathic pulmonary fibrosis (Thorax 52: 333, 1997). The response to steroids in this disease is generally good.
RESPIRATORY BRONCHIOLE-ASSOCIATED INTERSTITIAL LUNG DISEASE ("smokers' respiratory bronchiolitis"): Fibrosis primarily around the respiratory bronchioles, with plenty of macrophages nearby. A smoker's lesion likely to be picked up by a radiologist (AJR 173: 1617, 1999). Pathologists see Arch. Path. Lab. Med. 134: 134: 27, 2010. Mixed obstructive-restrictive pattern on pulmonary function tests of course.
NONSPECIFIC INTERSTITIAL PNEUMONITIS is a descriptive diagnosis that includes an entity that also responds well to glucocorticoids. It is distinguished from UIP / idiopathic pulmonary fibrosis by the uniformity of the histologic changes, with all septa involved about equally, and no "honeycomb cysts". Chest 125: 522, 2004. The idiopathic form usually affects middle-aged women who have never smoked, and the outcome is good (consensus document AJRCCM 177: 1338, 2008). Commonly (but by no means always) the "pulmonary fibrosis" in systemic autoimmune ("collagen-vascular") disease is of this pattern. Be this as it may, the prognosis is generally better than for classic idiopathic pulmonary fibrosis (Am. J. Resp. Crit. Care Med. 175: 705, 2007; Chest 134: 601, 2008).
![]() Desquamative interstitial pneumonitia
Desquamative interstitial pneumonitia
Lung pathology series
Dr. Warnock's Collection
RADIATION LUNG, AMIODARONE LUNG, CYTOXAN LUNG, BUSULFAN LUNG, BLEOMYCIN LUNG (Am. J. Path. 147: 352, 1995), and GVH-DISEASE LUNG: fibrosis due to cancer therapy
PARAQUAT INGESTION -- patients who drink this herbicide die of rapidly-developing, severe fibrosis of the lungs.
SARCOID LUNG: see below
POLYMYOSITIS-DERMATOMYOSITIS: Especially when antibodies against t-RNA synthetases (anti-Jo, etc.) are present (update Arth. Rheum. 56: 1295, 2007).
IDIOPATHIC PULMONARY HEMOSIDEROSIS: usually-mild, not-very-serious illness of young people with recurring microhemorrhages into the alveolar spaces
* ACUTE IDIOPATHIC PULMONARY HEMORRHAGE occurs in babies and can be fatal. It's presently being worked-out (MMWR 50: 494, June 15, 2001 -- three of the four index children had von Willebrand's; Am. J. For. Med. Path. 22: 188, 2001. There was a flap about about a mold in the homes of these children as the cause of a supposed epidemic in Cleveland (Pediatrics 99: E5, 1997 -- the MMWR folks found mold in these homes like everybody else's). I'm undecided.
* Angiosarcoma metastatic to the lung is a hard-to-diagnose cause of diffuse pulmonary hemorrhage: Arch. Path. Lab. Med. 125: 1562, 2001.
WEGENER'S you know. Pulmonary capillaritis has been described in anti-myeloperoxidase disease (Am. Rev. Resp. Dis. 146: 1326, 1993; the histopathology is the same as pulmonary Wegener's) and generally in systemic vasculitis (fibrinoid precedes neutrophils: Arch. Path. Lab. Med. 121: 144, 1997; Am. J. Clin. Path. 104: 7, 1995.)
|
|
|
|
|
|
|
|
|
Interstitial fibrosis is one component of ARDS and BRONCHOPULMONARY DYSPLASIA (the latter follows oxygen treatment of neonatal RDS)
DIFFUSE PULMONARY AMYLOIDOSIS, part of the systemic disease in a few cases
Nodular amyloidosis
Lung pathology series; follow the arrows
Dr. Warnock's Collection
ORGANIZING PNEUMONIA ("cryptogenic or secondary organizing pneumonia", formerly "BOOP"; update Am. J. Med. Sci. 335: 34, 2008)
This is a histopathologist's word for a lesion in which little pieces of loose connective tissue develop and plug the respiratory bronchioles and alveolar ducts and spaces. Air flow is obstructed, and lung expansion restricted. Although the lesions are intraluminal rather than interstitial, the spirometric pattern is more typical of a restrictive lung disease.
![]() Organizing pneumonia simulating coal dust disease
Organizing pneumonia simulating coal dust disease
Short paper by your instructor
For your enjoyment
This unhealthy situation, which mimics an acute pneumonia with a characteristic appearance on scan, can be seen in any of the following:
Very often, a short course of glucocorticoids will effectively treat "organizing pneumonia". The term "bronchiolitis obliterans organizing pneumonia / BOOP", though fun to say, has been discarded because of improved recognition of the completely-different and less-treatable entity obliterative bronchiolitis.
|
|
|
LYMPHANGIOLEIOMYOMATOSIS: a rare disease with marked proliferation of the smooth muscle throughout the lung.
* The cell of origin is claimed to be the "perivascular epithelioid cell", which lights up with HMB-45, as well as actin and desmin. The kidney tumor typical of tuberous sclerosis also stains for HMB-45.
* The genetics and molecular biology: Arch. Path. Lab. Med. 134: 33, 2010. The lesions may be widespread, or there may be one focal lesion, "lymphangio(leio)myoma"; imaging studies make the distinction.
* Future pathologists only: People with tuberous sclerosis also tend to have hyperplastic nodules made of type II pneumocytes.
* Sirolimus, the mTOR signal inhibitor, for lymphangioleiomyomatosis: NEJM 358: 140, 2008; NEJM 364: 1595, 2011.
* Future pathologists only: The benign "sugar tumor" of the lung, with cells loaded with glycogen, also is supposed to arise from perivascular epithelioid cells ("PEComa") and is HMB-45 positive.
![]() Lymphanioleiomyomatosis
Lymphanioleiomyomatosis
Lung pathology series
Dr. Warnock's Collection
![]() Pulmonary lymphangiomyomatosis
Pulmonary lymphangiomyomatosis
Great photos
Pittsburgh Pathology Cases
ALVEOLAR PROTEINOSIS: surfactant and proteinaceous goop fills the alveoli (same as in acute silicosis) -- listed here by "Robbins", but rarely includes fibrosis.
Most cases are idiopathic, and probably result from some loss of ability of the type II pneumocytes and/or alveolar macrophages to dispose of surfactant. Antibodies against GM-CSF, the macrophage colony growth factor, are known to be one cause.
Infectious agents (notably mycobacteria, but a host of others have been mentioned) are often present; no one knows whether they interfere with surfactant processing and/or simply grow well in this culture medium.
* Certain amphophilic drugs can induce alveolar proteinosis, perhaps by interacting with the soapy surfactant itself. Alveolar proteinosis caused by smoking fentanyl patches resolves when the person abandons the practice (Chest 141: 1321, 2012).
Several genetic forms involving mutated surfactant proteins have been discovered. The first was absent surfactant protein B (NEJM 328: 406, 1993).
By serendipity, a knockout mouse lacking GM-CSF (granulocyte-monocyte colony stimulating factory) turned out to have alveolar proteinosis, and many patients with the "idiopathic" form turn out to have autoantibodies against this factor (NEJM 356: 567, 2007; Am. J. Resp. Crit. Care Med. 177: 752, 2008) or genetic deficiencies in GM-CSF (Am. J. Resp. Crit. Care Med. 182: 1292, 2010).
Regardless of cause, bronchial lavage is the mainstay of therapy.
![]() Alveolar proteinosis from silicosis
Alveolar proteinosis from silicosis
Lung pathology series
Dr. Warnock's Collection
![]() Amyloid and proteinosis
Amyloid and proteinosis
Lung pathology series
Dr. Warnock's Collection
Don't forget CHRONIC HYPERSENSITIVITY PNEUMONITIS (Chest 134: 126, 2008; Am. J. Clin. Path. 131: 405, 2009; Am. J. Clin. Path. 134: 613, 2010), with or without an occupational history. Fibrosis is worst around the small bronchioles in the centers of the lobules; the apices tend to be involved worst; and there's usually some loose granulomas.
Cases in which the etiology is totally obscure are called IDIOPATHIC PULMONARY FIBROSIS / CRYPTOGENIC FIBROSING ALVEOLITIS. See below.
* NOTE: The "Loeffler's" family of eosinophilic pneumonias seldom produce fibrosis.
Clinicians: "Restrictive lung disease" (sort of the opposite of "obstructive lung disease") exists when the airways are widely patent but there is a problem ventilating the lungs. ("Restrictive" and "obstructive" lung disease give contrasting patterns on spirometry. Other causes of "restrictive lung disease" include large pleural effusions or cancers in the pleural spaces, pneumothorax, chest wall and spinal column problems, lungs full of tumor, anaconda attack, metastatic calcification, massive obesity -- don't overlook this one Am. J. Med. 116: 58, 2004-- etc, etc.)
IDIOPATHIC PULMONARY FIBROSIS / "cryptogenic (idiopathic) fibrosing alveolitis", "usual interstitial pneumonitis" and its kindred: Chest 128(5 S 1): 526-S, 2005; CMAJ 171: 153, 2004)
Pulmonary fibrosis, most often occurring in middle-aged and older people, and progressing to death.
"Usual interstitial pneumonia", or "UIP" is the most common and most deadly of several different processes in which pulmonary fibrosis appears. It is recognized by uneven fibrosis of the alveolar septa themselves, with overlying cuboidalization of the epithelium, and here and there, sites that look like healing injuries ("fibroblast foci").
At the end, the lung looks grossly like a coarse sponge, and feels like one only firmer. The alveoli are thickened, though some areas are always spared. Focal areas of hyperplastic fibroblasts ("fibroblastic foci" -- morphometry AJRCCM 174: 623, 2006 and Chest 130: 22, 2006) have overlying type II pneumocytes. Contraction of the fibrous tissue causes dilation of some of the air spaces ("honeycomb cysts").
The involved alveoli show obvious chronic inflammation, but no pathogenic micro-organism or other etiology has been forthcoming.
Idiopathic pulmonary fibrosis is one of several common illnesses with ongoing, unexplained, seemingly self-perpetuating inflammation. (* Similar mysteries include rheumatoid arthritis, Crohn's disease, and psoriasis.)
* In the 1990's, the focus was on altered alveolar macrophage function and overproduction of PDGF, interluekin 8, and so forth.
Today's work focuses on the complexity of the underlying immune abnormalities. All patients have abnormal T-cell (CD4+) clones, and the vast majority also have IgG antibodies against lung antigens (J. Imm. 179: 2592, 2007).
A familial syndrome exists; the gene is at the telomerase locus, making these patients victims of a forme-fruste of dyskeratosis congenita, which features a UIP-style pulmonary fibrosis often with marrow failure as well (NEJM 356: 1317, 2007; see also AJRCCM 178: 729, 2008 -- many idiopathic pulmonary fibrosis patients without mutations have short telomeres in their white blood cells for some unknown reason.) This now seems to be quite common (Blood 117: 5607, 2011)
* At least one family has a surfactant protein C protein mutation instead: AJRCCM 165: 1322, 2002). MUC5B promoter polymorphism: NEJM 364: 1503, 2011.
* Watch this one... are some (most?) UIP patients suffering a vicious cycle that begins with obstructive sleep apnea? The pathology community is starting to talk about this (Arch. Path. Lab. Med. 136: 470, 2012)
Your lecturer was even more impressed by the idea that idiopathic pulmonary fibrosis is a self-perpetuating tractional injury on the aging (elastin-poor) lung (Arch. Path. Lab. Med. 136: 591, 2012). It's based around 3D reconstructions of the fibroblast foci, which when connected look like the cracks in a dry river bed. (One sees fibroblast foci after recurrent spontaneous pneumothorax as well.)
Survival times are improving, with 5 years or longer now being common.
|
|
|
{40688} Idiopathic pulmonary fibrosis, gross, remember scar is white
{40685} Idiopathic pulmonary fibrosis, histology, the alveoli are slits
Glucocorticoids are notoriously ineffective for UIP, though they work better for its clinical mimics (desquamative, lymphoid, and nonspecific interstitial pneumonias). So far, no other medication has been shown to improve survival for UIP.
* There's an accelerated form (?) called "acute interstitial pneumonia". It follows a cold, gets worse over days or weeks, involves only the lungs, and looks just like ARDS under the microscope. There are reports of cures with glucocorticoids (Chest 124: 554, 2003).
* Famous "Idiopathic pulmonary fibrosis" victims include stunt rider Evel Knievel, actor Marlon Brando, and comic Jerry Lewis.
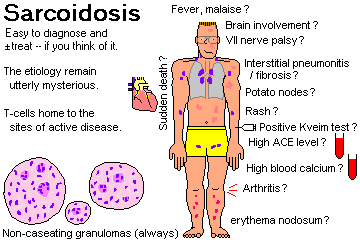
SARCOIDOSIS ("sarcoid", "Boeck's sarcoid"; * "sarcoid" literally means "the fleshy disease"): "A multi-system disorder of unknown etiology characterized by formation of noncaseating granulomas" (Baby Robbins) in many organs of the body, with variable clinical course. Reviews Lancet 361: 1111, 2003; JAMA 305: 391, 2011.
Sarcoidosis is probably an exaggerated ("hyperimmune") response by T1H lymphocytes to one or more unknown agents. CD4 lymphocytes home to involved areas, and macrophages form granulomas here. Sarcoidosis might even be a reaction pattern rather than one disease.
Most sarcoid patients are young adults. In the US, women and blacks are more often affected, but no group is immune. In about 2/3 of patients, the disease remits on its own.
There is only a slight familial tendency. * Despite the fact that the disease is common in US blacks, it's rare in sub-Saharan Africa.
The majority of people with sarcoidosis probably never become symptomatic. Those that do are likely to have:

Cardiac sarcoidosis is seldom recognized but lethal, especially when granulomas in the AV node cause 3rd-degree heart block and sudden death (CMAJ 136: 1064, 1987; Thorax 44: 371, 1989; Am. J. Card. 104: 571, 2009). Suspect it whenever a young person has heart block or focal echo abnormalities of the ventricular wall (Br. Heart. J. 57: 256, 1987). Any unexpected sudden death (even a drowning -- good swimmers and bath-takers don't "just drown") -- Am. J. For. Med. Path. 34: 11, 2013. Pathologists see Arch. Path Lab. Med. 134: 1039, 2010.
* In one variant of cardiac sarcoidosis, the heart is massively involved, with minimal involvement of other organs (Br. Med. J. 292: 1095). Update on the deadliness of sarcoidosis: Am. Heart. J. 150: 45, 2005.
The "cause" of sarcoidosis continues to elude us. Since the eyes and lungs are the areas most often involved, it's inviting to think it is a reaction to something we inhale.
* Mycobacteria, clay dust, and pine pollen have been suggested, but never demonstrated, as "antigens". More recently, interferon therapy has apparently caused "sarcoidosis": Cancer 59: 896, 1987, and it may appear in response to immune reconstitution in patients receiving HAART for HIV. Among firefighters inhaling dust after the World Trade Center disaster, there has been a great deal of apparent sarcoidosis, both in the lung and extrapulmonary (Chest 131: 1414, 2007).
The immune disturbance seems central.
There are lots of T4-helper cells at sites of inflammation, very few in the circulating blood.
Further, the body's B-cells are hyperactive (polyclonal gammopathy) -- it seems likely they are getting bad advice from the T4-cells.
Lymph nodes draining a cancer occasionally exhibit a morphologic reaction identical to sarcoidosis (Cancer 68: 1845, 1991) that may stay around after the cancer is cured (Chest 98: 1300, 1990).
* Sarcoidosis is famous for popping up in its victims' tattoos (Arch. Derm. 141: 869, 2005; NEJM 370: e34, 2014; JAMA 313: 1747, 2015).
Sarcoid granulomas are sharply circumscribed, tend to occur adjacent to and follow along lymphatic vessels, usually do not caseate, and contain giant cells. (You may hear that foreign-body giant cells are most characteristic of sarcoidosis, but actually Langhans cells are just as common.)
|
|
![]() Sarcoid
Sarcoid
Lymph node
Wikimedia Commons
{08756} sarcoid in the lung
{11417} sarcoid in a node, asteroid body
{11464} sarcoid, odd stain
{11774} sarcoid, lymph node
{12376} lupus pernio
{10976} sarcoid granuloma
{10979} sarcoid granuloma
{14427} sarcoid granuloma
{15474} brain sarcoid
{20220} sarcoid, lymph node
{23380} sarcoid, granulomas
{34913} sarcoid, lung
{35945} sarcoid, muscle
{38416} gross, sarcoid
{42030} sarcoid, marrow
* "Asteroid bodies" (probably wreckage from the cytoskeleton), "Schaumann's conchoid bodies" (laminated calcium and iron, refractile), and/or oxalate crystals (pathologists: don't mistake these for talc) can turn up inside the giant cells, but have no diagnostic utility.
The diagnosis of sarcoidosis is usually made by finding noncaseating granulomas on biopsy of some organ, in the absence of infectious organisms, foreign body, or other explanation.
* Every writer on sarcoid seems to have a favorite organ to biopsy first. Right scalene node is standard, but you'll hear about biopsy of the lip (minor salivary gland), liver, bone marrow, and even fine-needle aspiration of the spleen, all in a search of noncaseating granulomas.
Serum angiotensin converting enzyme (ACE) is a useful adjunct test (elevated more often than not), to make the diagnosis and monitor disease activity.
You'll hear about the Kveim (Kveim-Siltzbach) test, which involves taking ground-up spleen from someone with sarcoidosis and injecting it into the dermis. If a granuloma forms, the living patient supposedly has sarcoidosis (80% sensitive, 95% specific). Long considered outdated, there is now a resurgence of interest in the material, especially among researchers who need to decide who really does and does not have the disease (Clin. Chest Med. 18:799, 1997; Mount Sinai Journal of Medicine 63:335, 1996; J, Imm. 154: 1450, 1995). To date, nobody knows what's in Kveim reagent, except that it's supposedly not bacteria: AJRCCM 159 1981, 1999; Exp. Lung Res. 30: 181, 2004.
Making the diagnosis is important, because immunosuppression using a few weeks on glucocorticoids is often helpful. Longer therapy is much more dubious; the side-effects of the drugs are noxious, and you're not going to clear up scar tissue.
* Sarcoidosis is a common, lethal disease in horses. Oddly, amyloidosis almost never complicates sarcoidosis (Thorax 43: 422, 1988).
* Treatment of cutaneous sarcoidosis using tetracycline: Impressive. Is this really a bacterial disease after all? Arch. Derm. 137: 69, 2001.
* "Giant cell interstitial pneumonitis" is a recognizable reaction pattern almost always caused by inhalation of hard metal dust (see above; reaffirmed AJRCCM 176: 834, 2007). The give-away is the presence of uninucleate macrophages engulfed within giant cells.
* Whether or not "Blau syndrome", a childhood genetic disease at NOD2 which mimics sarcoidosis, will prove to be related remains unknown: Arch. Derm. 143: 386, 2007.
GOODPASTURE'S DISEASE: Antibodies against the basement membranes of lung and kidneys -- type II immune injury.
In Goodpasture's, pulmonary hemorrhage ranges from mild to exsanguination / drowning in blood. We will see more of this when we talk about "Kidney" -- these patients die of renal involvement.
Not all of these patients have renal involvement clinically: Thorax 46: 68, 1991.
{24848} Goodpasture's, lung
{29104} Goodpasture's, lung, iron stain
{29581} Goodpasture's
{29584} Goodpasture's
![]() Lupus and Goodpasture's
Lupus and Goodpasture's
Lung pathology series
Dr. Warnock's Collection
EOSINOPHILIC PNEUMONIAS ("pulmonary eosinophilia", "Loeffler's", etc.; Med. Clin. N.A. 95: 1163, 2011)
A grab-bag of illnesses ("idiopathic", drug-related, myeloproliferative -- famously chronic eosinophilic leukemia -- and some are caused by worms) that feature many eosinophils in the blood and sputum and in the pulmonary alveolar walls. Patients usually have pulmonary infiltrates and increased eosinophils in the blood.
"Simple Loeffler's" ("acute eosinophilic pneumonia", a febrile illness with pulmonary infiltrates which if biopsied prove rich in eosinophils) probably gets missed most of the time, presenting and behaving as a simple "chest cold." A known cause is starting to smoke (Lsncet 385: 1150, 2015), or restarting, or increasing one's frequency (Chest 133: 1174, 2008).
In the U.S., the longstanding disease is most often due to aspergillus![]() colonizing the airways of an asthmatic. But
there are many other causes, including ascaris worms migrating
through the lungs, and plenty of
"idiopathic" cases.
colonizing the airways of an asthmatic. But
there are many other causes, including ascaris worms migrating
through the lungs, and plenty of
"idiopathic" cases.
In "tropical eosinophilia", microfilaria worms are trapped in the lungs and break down, producing a similar syndrome.
* While we are on the subject of worms in the lungs, remember that dirofilariasis, the dog heartworm, is prone to die in human lungs and produce a "coin lesion" simulating lung cancer. I suspect this accounts for a few cases of "surgically cured squamous cell carcinoma of the lung."
|
|
Among drugs, the best-known offender is nitrofurantoin.
Some of these patients will have Loeffler's eosinophilic endocarditis.
* Meprolizumab, the interleukin-5 inhibitor, is an effective and well-tolerated way to control the wheezing in eosinophilic pneumonia (Lancet 380: 651, 2012).
* Churg-Strauss vasculitis may feature an "eosinophilic pneumonia" -- future pathologists, look hard for granulomas before making your diagnosis of "idiopathic Loeffler's" (Am. J. Clin. Path. 114: 767, 2000). Remember that these patients have asthma, often "new onset", ANCA is usually positive in these patients.
Whatever the hypereosinophilic diseases (respiratory system, GI tract, hematopoietic) really are, the availability of mepolizumab (anti-interluekin-5 antibody) will probably prove helpful both in treating them and in discovering their real nature (J. Allerg. Clin. Imm. 118: 1312, 2006; NEJM 358: 1215, 2008).
Do not confuse these with EOSINOPHILIC GRANULOMA, a member of the "histiocytosis X" family of quasi-neoplastic proliferations of Langerhans histiocytes, seen mostly in middle-aged smokers.
|
|
|
LIPID PNEUMONIA
{38464} lipid pneumonia
|
EXOGENOUS LIPID PNEUMONIA: the lung's reaction to aspirated oil.
Causes include oily nose drops, mineral-oil laxatives, and forcing children to swallow cod-liver oil (the latter must have been an important killer of children, whose pitiful deaths were due to "pneumonia"). Don't give this stuff to your patients.
The more unsaturated the oil, the worse the inflammation. Mineral oil disease is usually mild. Polyunsaturated vegetable oils are worst.
The pathologist sees yellow patches grossly. The microscopic picture features lipid-laden macrophages and fibroblasts laying down scar tissue.
* "Fire eater's pneumonia" is actually due to aspiration of petroleum products, though it mimics an infection clinically (Chest 124: 398, 2003).
ENDOGENOUS LIPID PNEUMONIA ("golden pneumonia" / "(post)-obstructive pneumonia"): buildup of surfactant (in macrophages) behind an obstructed major airway (i.e., lung cancer) or many small airways ("obliterative bronchiolitis" family), or in amiodarone lung (as a phospholipase inhibitor, it interferes with surfactant processing: Chest 112: 1068, 1997).
|
|
|
![]() Amiodarone lung
Amiodarone lung
Lung pathology series
Dr. Warnock's Collection
* MISCELLANEOUS LESIONS
BRONCHOCENTRIC GRANULOMATOSIS: A reaction pattern, most
often seen with infection by
Aspergillus fungus![]() ,
in which the bronchi break down into
caseating
,
in which the bronchi break down into
caseating![]() granulomas.
granulomas.
INVASIVE ASPERGILLOSIS throughout the lung is a dread infection of people with severe neutropenia. It's hard to diagnose, even with a couple of blood tests that detect fungal chemicals.
The CYSTIC ADENOMATOID LESION is a fortunately rare and dangerous hamartoma seen in infants.
A BROCHOPULMONARY SEQUESTRUM is a portion of lung tissue, inside or outside the healthy lung, with airways unconnected to the main bronchial tree, and therefore unaerated. It is prone to become infected, or to cause problems because of its mass.
PULMONARY INTERLOBAR SEQUESTRUM is a birth defect in which part of the lung is supplied by a branch of the aorta instead of by the pulmonary artery. This is only a problem later when it gets infected.
Several types of CONGENITAL CYSTS OF THE RESPIRATORY TREE also occur and can cause problems (hemorrhage, infection, rupture, etc.) at any age.
Remember that most lung diseases will be exacerbated by gastric reflux.
|
|
|
|
|
Future pathologists: See Arch. Path. Lab. Med. 133: 1106, 2009! Non-small-cell: Lancet 378: 1727, 2011; small-cell Lancet 378: 1756, 2011. Clinicians CA 61: 91, 2011. How pathologists approach the molecular diagnosis today: Am. J. Clin. Path. 138: 332, 2012; Arch. Path. Lab. Med. 137: 481 & 1191 & 1274, 2013.
The vast majority of primary malignant neoplasms of the lungs are some form of BRONCHOGENIC CARCINOMA.
The "million reported deaths each year worldwide" (NEJM 356: 830, 2007; 1.5 million Lancet 382: 709, 2013) seems way-low in a world where Russia, China, and many of the poor nations have much higher rates of smoking than the US but where smoking kills maybe 160,000 people yearly.
In 2011, there were around 226,160 new cases in the US, and 160,340 deaths. The total number of cases has increased greatly in the last decade because we are finding cases earlier using imaging; however the total number of deaths has dropped only modestly.
Bronchogenic carcinoma is the leading cancer killer of US men and women. Age-adjusted rates have been dropping for men since 1980 (NEJM 321: 1197, 1989), and the total absolute number is now decreasing; the total percent of the overall US population getting sick from / dying of lung cancer is way down.
It surpassed breast cancer as the leading cancer killer of women in 1985. The rates in women have started to drop (2004).
Still, the majority of lung cancers are silent until they've become inoperable. Those lucky patients whose cancers are of operable subtype and are detected very early have a reasonably good prognosis (maybe 60% long survival with stage I, 40% stage II: Ann. Thorac. Surg. 60: 466, 1995).
Lung cancer exhibits Nowell's law in action, with mutations in the surrounding "normal" epithelium. Prognosticating lung cancer using genetic markers has been underway since the mid-1990's, and folks who study the various mutations now look at around 100 genes (Am. J. Resp. CCM. 170: 167, 2004).
|
Risk factors are well-established (Chest 103-S1: 20-S, 1993) CIGARET |
 |
The danger is proportional to the amount of smoke inhaled every day and the duration of smoking, and is measured in "pack-years." After ten pack-years the increased risk is measurable, and after fifteen pack-years, there is clear-and-present danger. After quitting, the risk decreases slowly and approaches normal after ten or fifteen years. (Cigar smokers inhale less smoke overall than cigaret smokers, but contrary to what you may hear, their risk for lung cancer is still greatly increased.)
"Passive smoking" seems to be of some importance, but much less than active smoking (Am. J. Pub. Health 82: 1525, 1992). Pipe smoking and tobacco chewing are much less risky than cigaret smoking, at least as far as the lungs are concerned. Snuff, for example, simply doesn't give Swedes lung cancer (Lancet 369: 2010, 2007).
The other risk factors are synergistic (i.e., more than just additive) with cigaret smoking.
* By the way, former smokers filling out research forms will often say they never smoked. Remember this when you're evaluating research articles (Chest 101: 19, 1992). In particular, every single article on "the relative risk of this-or-that controlling for smoking" is severely tainted for this reason alone.
RADIATION: uranium miners (powerful effect, all histologies Cancer 89: 2613, 2000), atomic bomb survivors, after radiation therapy (mild effect; Cancer 71: 3054, 1993), radon in homes ("if you say so....": NEJM 330: 159, 1994.)
* During the 1980's and early 1990's, there was a much excitement over radon from the ground building up in our energy-efficient homes. The claim, advanced by the Environmental Protection Agency, was that it caused 5-20 thousand lung cancers yearly, multiplicative with tobacco smoking. Was it just junk science? I wasn't the only one to say so at the time... Lancet 337: 1329, 1991; Arch. Int. Med. 151: 674, 1991 (AMA council report). Since "radon-proofing your home" was a mega-bucks business, relating to property values and fear of government regulation, someone more cynical than myself might have seen an even baser motivation.
* It does seem possible that smokers are at increased risk if there's radon in their homes. You read up and decide. Most of the epidemiologic studies came back negative, one controversial study found a negative correlation (as if radon protected you: Health Phys. 72: 623, 1997), the last big paper to find a link was Am. J. Pub. Health 89: 1042, 1999 and even the authors admit it's really weak, and even Br. J. Cancer 84: 134, 2001, which did a study of radon and lung-cancer in nonsmoking men, couldn't quite get a statistically significant correlation. There seems to be agreement that non-smokers aren't at serious risk, and that smokers can protect themselves much less expensively by stopping smoking (Am. J. Pub. Health 88: 811, 1998) -- or if Uncle Sam must mandate it, helping people stop smoking is a much more cost-effective way of controlling the harmful effects of radon than forcing people to spend megabucks to radon-proof their homes (Am. J. Pub. Health 103: 443, 2013). The collaborative analysis in BMJ 330: 223, 2005, including old and new studies only found a modest risk and then only for smokers or recent ex-smokers. Similar findings, including a statement that it simply isn't economically feasable even in a rich nation to reduce the risk by "radon-proofing" homes: BMJ 338: a3110, 2009.
As you would expect, the nucleic acid damage from radiation is more random than in tobacco smokers. See Lancet 339: 576, 1992.
Radon resurgent: In highly-polluted, no-EPA China, occupational radon exposure seems to correlate with atypical cells in the sputum. See Chest 135: 778, 2009. How to sort out co-existing variables seems problematic (are radon-exposed workers "told not to smoke" and therefore lie about it? are there other exposures for these unfortunates as well?)
ASBESTOS INHALATION: Notorious cause of lung cancer. According to some accounts, more than half of asbestos workers who smoke have died of lung cancer (more: Br. Med. J. 306: 1503, 1993).
* CERTAIN METAL DUSTS (supposedly nickel -- the major work is from the late 1970's and is small-sample statistics -- and maybe silver)
* HEXAVALENT CHROMIUM (the most familiar form in industry; the best study is J. Occ. Env. Med. 48: 426, 2006 which found some increased risk in the more heavily exposed German chromate workers when we control for smoking. The authors believed there was a threshold effect below which there was no measurable risk, which makes the activist-driven OSHA crackdown in 2007 senseless.
COAL TAR FUMES, carbon black (Lancet 358: 562, 2001) and other chemicals (* notably vinyl chloride, chloromethyl ethers) Occupational lung cancer: Mayo Clin. Proc. 68: 183, 1993.
URBAN POLLUTION: a minor risk factor compared with smoking / uranium mining / asbestos exposure, with which it may be synergistic (JAMA 287: 1132, 2002)
* In one study that made many people upset, the investigators failed to take into account the fact that people living near the steel mill smoked much, much more than did their rural counterparts. When somebody noticed this, the "cancer risk" from the steel mill disappeared (Arch. Env. Health 48: 184, 1993).
In the poor nations, living in a coal-heated dwelling that's not ventilated clearly causes a great deal of lung cancer (Lancet 362: 849, 2003; JNCI 94: 826, 2002; BMJ 345: e5414, 2012).
* I think that silica / sand exposure, made "official" in 1997 by the International Agency for Research on Cancer, is without any scientific merit. The numbers are very soft and there's no credible mechanism. See Am. J. Epidemiol. 153: 695, 2001.
* Red meat: The NCI does an 8-year study and finds a slight positive correlation between how well-done smokers like their meat and how much lung cancer they get: Am. J. Clin. Nutr. 89: 1884, 2009. That's all I could find and I'd caution anyone from drawing any unjustified conclusions.
|
 |
* For a while there was a hoopla about carotenoids (there are at least five kinds) as protecting people (smokers, non-smokers) from lung cancer. Given that a high level of blood carotenoids strongly suggests an overall healthy-lifestyle, and that today's "pop wisdom" is that vegetables protect from cancer, it's hard to do reliable studies. Protection by dietary carotenoids: Am. J. Clin. Nutr. 72: 990, 2000; Am. J. Ep. 156: 536, 2002 (about a whopping 15%, and based on self-reports); no protection Canc. Causes Contr. 13: 231, 2002; more confusion Br. J. Cancer 84: 728, 2001 (something's going on but it can't all be the tomato sauce).
Familial cancer syndromes (i.e., antioncogene deletion syndromes) causing lung cancer in non-smokers are conspicuous by their absence. Li-Fraumeni and retinoblastoma-family syndromes give increased risk if you smoke. A susceptibility gene (point-mutation) discovered in 2008 turned out to be (surprise!) the nicotine receptor (Nature 452: 633, 2008; Nat. Genet. 40: 616, 2008 -- and the mechanism is ... you smoke more cigarets! Nature 452: 638, 2008).
NOTE: We know who is at risk. The traditional wisdom for decades was that screening (routine x-rays, exfoliative cytology) was a waste of time and money (Ann. Int. Med. 111: 232 & 239, 1989). With today's improved low-radiation scanning, we have begun puting smokers into the machine yearly: Cancer 89(S-11): 2474, 2000. The initial excitement ("We're curing 60-80% of the early cancers we find!") gave way to disappointment with the lack of drops in overall cancer deaths -- perhaps we're finding only the very slow-growing cancers. Given the cost, inconvenience and dangers of the workup for a lung spot, today there's no consensus as to whether to scan smokers routinely (JAMA 309: 1163, 2013). Even the JAMA, not known as a bastion of noninterventionism, is coming around to my idea that the cancers we're finding and curing on intensive screening are the ones that wouldn't have killed the person in any case: JAMA 311: 349, 2014. "$81K per quality adjusted life year gained" NEJM 371: 1793, 2014. Helpful or not, screening of folks age 55-74 with 30+ pack-yearsis now paid for by Medicare in the belief that lives are indeed saved (NEJM 372: 2083, 2015 -- NLST; National Lung Screening Trial).
Classification scheme:
The WHO classification was updated in 1999 and again in 2004. Most bronchogenic carcinomas still get assigned to one of the four light-microscopic categories of the old WHO classification (1967):
LUNG CANCER DIAGNOSIS FOR BEGINNERS (finding any item in the group usually makes the diagnosis...)
Squamous Cell...keratin, squamous pearls, "bridges" (desmosomes); EM: tonofilaments, diffuse keratin; immunoperoxidase stain for high-molecular-weight keratin or p63, p40, cytokeratin (CK) 5/6, DG3 (desmoglein 3)
* As above, TTF-1 is usually negative; this may become part of the definition someday.
Today, an oncologist or pathologist may define a non-small-cell lung cancer to be squamous if and only if it lights up with p63.
Adenocarcinoma... glands, papillae, mucin; EM: microvilli, secretory granules, gland formation; IP: low MW keratin, CEA; napsin-a; TTF-1 (the latter is especially helpful as an indicator that an adenocarcinoma metastasis of unknown origin came from lung / thyroid / esophagus)
* Other adenocarcinoma subtypes exist and except as noted below are of no known significance.
* For the more obscure histologic types of adenocarcinoma, see Arch. Path. Lab. Med. 130: 958, 2006.
The larger the nuclei in an adenocarcinoma, the worst the prognosis (Cancer 116: 2011, 2010).
* The WHO prefers a common-sense grading system from 1982:
The newest proposed grading system ("Barletta") which supposedly predicts prognosis looks at (1) how much of the pattern is solid), and (2) how bizarre the cells are. It actually has strong predictive value (long vs. short survival time), and it's quite simple:
It does not apply to micropapillary or bronchioloalveolar tumors.
This is similar to the major grading system for breast adenocarcinomas. See Cancer 116: 659, 2010.
* Yet another study of histopathology: Classify them by growth pattern (lepidic, acinar, papillary, micropapillary, and solid). If there are any solid / sheets areas, the prognosis is worse. Cancer 118: 2889, 2012.
Large Cell Undifferentiated... none of the above, plenty of cytoplasm; the World Health Organization has added subtypes that need not concern us
Today, an oncologist or pathologist may define a non-small-cell lung cancer to be "not otherwise specified" (NSCLC-NOS) if it does not light up with either p63 or p40 or TTF-1. (Some oncologists will allow "Napsin A" positivity to make it an adenocarcinoma, and CK 5/6 to make an obvious non-mesothelioma a squamous cell carcinoma). There are still plenty of these "NSCLC-NOS"'s.
Small Cell Undifferentiated... "small blue cells" (i.e., very little cytoplasm to stain pink); EM: "neurosecretory" (dense-core, APUD) granules, fine chromatin, little cytoplasm; IP: neuron-specific enolase, bombesin, TTF-1 (Mod. Path. 19: 1117, 2006)
The trend still seems to be "men get more squamous and oat-cell cells, women get more adenocarcinomas" (Cancer 69: 86, 1992; Cancer 103: 2566, 2005; we very macho men cannot inhale our non-filtered cigs as deeply as the ladies inhale their filtered cigs).
More about these below. EM, immunohistochemistry, and genomics have further complicated things.
Nowadays, a lung mass is likely to be diagnosed by the pathologist performing fine-needle aspiration (AJRCCM 174: 684, 2006 -- a landmark study that shows that this does not cause tumor spread, as had been feared).
Until recently, clinicians considered it most important that the pathologist distinguish small cell undifferentiated carcinoma ("small cell" carcinoma) from the others. This is still true, but nowadays, anything that might be an adenocarcinoma will get some molecular subtyping anyway since there are some chemotherapeutic agents that a reasonable patient might accept and an ethical oncologist might use.
Today, an oncologist or pathologist may define a non-small-cell lung cancer to be adenocarcinoma if and only if it lights up with TTF-1. Or the pathologist may see one or more of the classic adenocarcinoma patterns and go right to gene markers.
* Resistance to crizotinib evolves after a while. One mechanism NEJM 368: 2395, 2013.
"Small cell" carcinoma has not been considered a surgical disease, and usually had a nice initial response to chemotherapy. See below.
And most recently, a Japanese team claims to have 7 of 9 patients operated for "stage I oat cell with negative lymph nodes" alive five years later (Ann. Thorac. Surg. 90: 229, 2010).
The other types have historically been treated surgically if feasible, radiated ("spot-welded") as needed, and occasionally responded well to non-targeted chemotherapy. For stage I adenocarcinomas resected for cure, about 2/3 stay cured (Ann. Thor. Surg. 90: 1067, 2010).
Chemotherapy for lung cancer was a discouraging business until recently. My own favorite article from 1998 was BMJ 317: 771, 1998: People who had been given chemotherapy for advanced non-oat-cell carcinoma of the lung would NOT have accepted it if they'd been told the truth beforehand. And a comparison of four expensive, horrific protocols that showed that none gave better than 20% transient responses ended with the frank admission that it's was wrong to do this (NEJM 346: 92, 2002).
During the first few years of the 21st century, treatment outcomes for non-oat-cell lung cancer became a bit better using platinum-based chemotherapy ("we''ve reached a plateau": Chest 136: 1112, 2009).
When the new VEGF inhibitor bevacizumab was introduced, it helped some people but famously caused massive, fatal hemorrhage in squamous cell carcinoma (which typically kills by hemorrhage anyway -- the bevacizumab just speeded it up).
The epidermal growth factor receptor, coded by the EGFR / ErfB-1 / HER1 proto-oncogene (Nobel Prize 1986), is often much over-expressed in non-oat-cell lung cancers (Nature 307: 521, 1984). This used to be just a way of telling oat-cell from non-oat-cell, but things recently got interesting.
Erlotinib ("Tarceva") has replaced gefitinib in the US; unlike its predecessor, it does seem to prolong survival and perhaps quality of life. Again, resistance develops in a few months.
The most promising study, which is now standard-of-care, found that patients with some (not all) mutations in EGRF (famously women who never smoked and had adenocarcinomas) got quite good temporary results with erlotinib (NEJM 361: 958, 2009; Arch. Path. Lab. Med. 133: 470, 2009).
There are three ways in which a tumor becomes resistant to EGFR inhibitors (Arch. Path. Lab. Med. 136: 1205, 2012)
The T790M mutation in EGFR kinase is a sign of developing resistance to tyrosine kinase inhibitors. The group watched their patients develop resistance to "Iressa" and "Tarceva" by monitoring EGFR mutations in the circulating blood (NEJM 359: 366, 2008).
Or the tumor can develop amplified MET/HGFR, which bypasses EGFR.
Or the tumor can be overgrown by, and turn into, oat cell carcinoma, which isn't driven by EGFR.
* When resistance to the common EGFR inhibitors develops, some new agents (NEJM 372: 1689 & 1700 & 1760, 2015) may still work.
* More biotech products are in the pipeline. Beware -- hopes for effective treatment of our great cancer killer have attracted crooks. The original article on the EGFR receptor (NEJM 354: 1176, 2006) led to a $1000 test that supposedly predicted whether a cancer would respond. It turns out test results didn't correlate well with outcome (Arch. Path. Lab. Med. 132: 1573, 2008), and the article was retracted because the investigators couldn't reproduce their own work (NEJM 364: 1176, 2011). You can read on your own and decide for yourself about Anil Potti. Stay tuned.
* ERCC1, an enzyme that does nucleotide excision repair of damaged DNA, may be checked in a tumor, with high levels indicating a better prognosis untreated but unresponsiveness to platinum-based chemotherapy (NEJM 355: 983, 2008).
Most encouraging, some new immune-system helpers give improved survival in non-small-cell lung cancers. Among the promising new agents is pembrolizumab, which inhibits a programmed-death ligand that's often seen on cancers -- a stain predicts response (NEJM 372: 2018, 2015)
* There are still some basic unknowns. For example, the "T"-stage (and prognosis, it seems) depends to a great extent on whether the tumor is invading through the visceral pleura, and there's no consensus among pathologists how to determine this; requiring clear penetration of the visceral elastic layer seems reasonable. (Am. J. Clin. Path. 128: 638, 2007; update Arch. Path. Lab. Med. 136: 1194, 2012.)
* My old friend Barrie R. Callileth and the rest of the integrative medicine team share an no-BadScience update on complementary therapies for the lung cancer patient, distinguishing the cynical frauds that pretend to affect the natural course of the disease to a host of treatments that can probably make people happier and more comfortable (Chest 132(S-3): 340-S, 2007).
|
|
SQUAMOUS CELL CARCINOMA ("Epidermoid carcinoma")
|
|
|
|
|
|
|
|
{17520} squamous cell carcinoma of lung,
large mass at hilum
{20994} squamous cell carcinoma of lung,
find the cavity
{20996} squamous cell carcinoma of lung,
gross
{17525} squamous cell carcinoma of lung,
poorly differentiated
{15401} tonofilaments
{15402} desmosome
You're already familiar with keratin pearls, desmosomes / prickles / bridges, and individual apoptotic cells. Today, CK 5/6, p63, p40, DG3 and absent TTF-1 help make the call.
Tobacco smoking is the major risk factor (90-99%, estimates vary).
Tumors arise anywhere in the bronchi, often near the hilum. Large squamous cell carcinomas tend to cavitate. Often hemorrhage into the cavity and out the mouth is the final event (exsanguination, drowning in blood).
Generally the pathologist can find squamous metaplasia and dysplasia (and even carcinoma in situ) in the bronchi surrounding a squamous carcinoma. ("Nowell's law" triumphant.)
Squamous cell carcinomas very commonly produce a parathyroid-hormone-like substance that may cause elevated serum calcium even in the absence of bone metastases -- "humoral hypercalcemia of malignancy". (Production of other ectopic hormones is unusual.)
The protein is PTHrP, parathormone-related peptide, required for proper development of teeth and bones (Proc. Nat. Acad. Sci. 95: 11846, 1998).
If the tumor is found when small, and is well-differentiated, the surgical cure rate is reasonably good. Squamous differentiation still remains an independent "good" prognositic sign in a bad disease: Ann. Thor. Surg. 77: 1173, 2004.
* Spontaneous regression of this dread cancer is very rare but happens: Chest 94: 701, 1988.
* Basaloid squamous lung cancer is a less-aggressive variant of squamous cell carcinoma recognizable by is histologic resemblance to basal cell skin cancer. Am. J. Clin. Path. 136: 436, 2011.
* Future pathologists only: The one benign fooler on histopathology is "necrotizing sialometaplasia". Everybody else: Don't worry about it.
* Two tumor markers for squamous cell carcinoma exist. To follow a patient with pulmonary squamous cell carcinoma for recurrence, you may use a serum SCCA ("squamous cell carcinoma antigen") or CYFRA21-1 ("cytokeratin 19 fragment"). Both of these have generated some interest.
|
ADENOCARCINOMA
|
|
|
You are already familiar with the common histology patterns that define adenocarcinoma. Today's pathologists may also make the call of adenocarcinoma if a tumor is strongly positive for TTF-1 or "napsin A" (non-mucus-producers, see Am. J. Clin. Path. 139: 160, 2013) or * PE-10.
Adenocarcinoma of the lung was once thought not to be related to tobacco smoking (Acta Path. Microbiol. Scand. S-157: 1, 1962.) Nowadays 75-90% of these patients in recent studies have been smokers.
Most adenocarcinomas arise in the periphery beneath or near the pleura.
|
|
|
|
{20997} peripheral adenocarcinoma
Adenocarcinomas have been said to arise in scars in the lung,
including old TB![]() , old infarcts, and physical wounds.
These are called "Yokoo tumors", and Dr. Yokoo taught me in residency.
I used to think that the
cancer produces the fibrosis, but I've seen several
adenocarcinomas at autopsy in non-smokers and these have always
been in the setting of scars of known etiology.
, old infarcts, and physical wounds.
These are called "Yokoo tumors", and Dr. Yokoo taught me in residency.
I used to think that the
cancer produces the fibrosis, but I've seen several
adenocarcinomas at autopsy in non-smokers and these have always
been in the setting of scars of known etiology.
The histology correlates with the prognosis for small tumors (Cancer 61: 2083, 1988 found that marked anaplasia and areas of solid growth are ominous, confirming today's data), while large tumors are very lethal.
* Surfactant apoprotein stain was an old stain that lit up many of these tumors and confirmed they were primary in the lung.
About 10% (figures vary) of adenocarcinomas are of the BRONCHIOLO-ALVEOLAR CARCINOMA subtype, considered to include cancers of the bronchial goblet cells ("mucin-positive", often K-ras-mutated) as well as type II pneumocytes and Clara / club cells ("mucin-negative"; includes the "sclerosing" sub-subtype).
These malignant cells grow along the alveolar septal framework and (if "mucin positive") secrete mucus into the alveoli, producing "consolidation." This cancer by definition does not invade, but kills by growing as a single layer of thick cells covering over the respiratory membranes.
This type of cancer is getting to be more common, or at least more recognized (Cancer 68: 1973, 1991). WHO-1999 separated it from other adenocarcinomas; WHO-2004 tossed it back in as a subtype. Before 1980 it was thought to be non-cigaret-related; newer studies show a relationship (but less than any of the four "major" types.)
* Pathologists distinguish primary bronchioloalveolar cell carcinomas from metastatic colon cancer (which can look identical) because only the latter stains for the villin antigen; this is not 100%. New technique: Cdx2 stain usually does not stain bronchiolo-alveolar carcinomas, but usually does stain colon metastases (Am. J. Clin. Path. 122: 421, 2004).
Beyond this, bronchioloalveolar carciomas are a genetically and immunologically heterogeneous lot: Arch. Path. Lab. Med. 128: 406, 2004.
Since a "pure" bronchioloalveolar carcinoma shows no evidence of stromal invasion, it's best considered "adenocarcinoma in situ", and some people are calling it that. Today, this is the common lung cancer "found on a screening CT scan and cured surgically."
* Future pathologists: the term for this pattern of growth along a surface without invasion is "lepidic".
|
|
|
|
|
|
|
|
|
|
|
{11454} bronchiolo-alveolar carcinoma; very
small cells growing along the alveolar septa
{12608} bronchiolo-alveolar carcinoma; the
lung is solidified by mucus in the alveoli
The tumor is slow-growing but very lethal unless it's a small resectable lesion (Ann. Thor. Surg. 71: 971, 2001). A few may be benign, but no one knows how to recognize these.
* Jaagziekte, an infectious disease of South African & other sheep, resembles this tumor histologically and clinically, but there is no good evidence of transmissibility in humans.
* Cigaret-smoking beagles, an animal model for human lung cancer, get mostly bronchioloalveolar carcinoma, and so do scleroderma patients (Arch. Pathol. Lab. Med. 108: 7, 1984.)
* It is worthless as a screening test, but to follow a patient with pulmonary adenocarcinoma for recurrence, you might use a serum CEA.
* Bronchioloalveoliar cell carcinoma patients who have their disease detected extremely early are the only lung cancer patients who may be offered lung transplants; around 50% are alive at five years (Ann. Thorac. Surg. 94: 935, 2012.)
* In 2011, a new system was introduced for classifying adenocarcinoma of the lung which will ERS (European Respiratory Society) -- see J. Thorac. Onc. 6: 244, 2011. probably become standard -- the IASLC (Intl. Assoc. Study of Lung Cancer) / ATS (American Thoracic Society) /
LARGE CELL UNDIFFERENTIATED CARCINOMA ("hundred-day cancer"; "non-small-cell carcinoma not otherwise specified")
|
|
|
|
|
|
|
Histologically, these tumors are made of big cells with plenty of cytoplasm and show no features of either adenocarcinoma or squamous cell carcinoma on H&E. Today, some will be defined as "squamous carcinoma" or "adenocarcinoma" if they show one of the markers listed above.
Most are related to tobacco smoking (90-99%, estimates vary.) These tumors arise anywhere in lungs. Cures are rare, and death comes quickly.
Obviously these tumors are mostly occupied with growth and invasion rather than looking like they did in health. There are no useful tumor markers (hPTH-like substance may be produced) or other clinical behaviors that really show this is a distinct entity.
SMALL-CELL UNDIFFERENTIATED CARCINOMA ("OAT CELL") (Lancet 378: 1741, 2011)
|
|
![]() Oat cell carcinoma
Oat cell carcinoma
These actually look like oats
Wikimedia Commons
{11428} oat cell carcinoma, spreading along
the bronchi
{10475} oat cell carcinoma, growing
down between the cartilage rings
{10435} oat cell carcinoma
{11693} oat cell carcinoma
{12578} oat cell carcinoma
{14076} oat cells, spinal fluid
{12581} oat cells in sputum
{38521} oat cell
{38527} oat cell
{09082} oat cell carcinoma, electron
micrograph showing neurosecretory granules
The "cell of origin" is the Kulchitsky (APUD) cell of the bronchial epithelium.
Almost all (99+%) tumors are related to tobacco smoking. (A few uranium miners with oat cell carcinoma claim not to have smoked.)
Small cell carcinomas arise anywhere in the lung, most often near the hilum, and quickly spread along bronchi.
This cancer is infamous for early and widespread metastases and rapid death of the patient. (Surgery is usually considered futile, but see above for very early lesions and decide yourself.)
The tumor is composed of small (2x size of a lymphocyte) cells with very little cytoplasm ("small blue cells", "oat cells"), with many mitoses and usually a lot of necrosis. The histology is so familiar that pathologists usually diagnose it with confidence without any ancillary studies; however, sometimes we order up a few special studies just to be sure.
The cells usually stain for TTF-1, p16(INK4A), neuron-specific enolase and the neuroendocrine markers CD56, synaptophysin, and others. EM shows oval nuclei, finely dispersed chromatin, scanty cytoplasm, usually a few neurosecretory (dense-core, APUD) granules.
Future pathologists: The tumor cells stain positive for neuron-specific enolase (NSE) and bombesin/GRP. While the three other major lung cancers usually stain for erbB, oat cell doesn't.
Like the K-cells from which they derive, "small cell" carcinoma is also known for secreting a lot of different substances.
Among these are ACTH, hADH, neurophysin, neuron specific enolase, bombesin (and/or the closely-related gastrin-releasing peptide), * bradykinin, * calcitonin, * growth hormone (hGH), * histamine, * hLH, * lipotropin (finding this suggests that elevated ACTH is not of pituitary origin: J. Clin. Endo. Metab. 86: 2997, 2001), * oxytocin, * prolactin, * somatostatin, * several different enzymes, etc., etc. (Bombesin/GRP is the cancer's autocrine growth factor).
IMPORTANT: Of these, the ones most likely to be detected are hADH (hyponatremia / water intoxication) and ACTH (Cushing's syndrome). Each syndrome occurs in 5-10% of "small-cell" patients (and ectopic secretion of the hormone is much commoner); they are seldom very serious but help make the diagnosis.
PTH-like hormone secretion and hypercalcemia are not typical of small-cell tumor patients.
* The molecular biology of small cell carcinoma has been under study since the early 1980's, without any signature genes being found. Best-known are amplifications of various myc genes. Update NEJM 359: 1369, 2008.
* Maybe 30% of oat cell carcinomas have at least a few little areas of apparent squamous cell carcinoma, adenocarinoma, or large-cell neuroendocrine carcinoma differentiation. These areas have pretty much the same mutations as the oat-cell cancer and the finding is of no known significance; likewise, if any lung cancer has any area of differentiation as an oat cell carcinoma, it should probably be considered as such (Am. J. Clin. Path. 131: 376, 2009).
Oat cell's initial response to chemotherapy is usually very good. Most cases relapse and die, but occasional cures of the early disease are now being claimed (Chest 123: 259-s, 2003; two percent of all patients alive at 5 years Cancer 89: 523, 2000); newer reports have up to 50% of patients who have very small oat-cell carcinomas at presentation alive at five years. Second primaries, recurrences and chemotherapy-induced leukemias may still occur and kill the patient.
* Future pathologists: (1) On biopsy, it's often tough to tell crushed oat cells from crushed lymphocytes, especially if your biopsy instrument is dull. Most helpful is a common leukocyte antigen (Semin. Onc. 20: 153, 1993). (2) On fine needle aspiration, there is a tendency to mistake oat-cell carcinoma for non-oat-cell carcinoma and carcinoids for oat-cell carcinoma. How not to make these errors: Arch. Path. Lab. Med. 129: 614 & 619, 2005.
* It is worthless as a screening test, but to follow a patient with pulmonary oat cell carcinoma for recurrence, you might use a serum neuron-specific enolase or progastrin-releaseing peptide (ProGRP). These are infamously nonspecific and nonsensitive.
* Quitting smoking after lung cancer has been discovered would seem futile, but it seem to give some increase in survival (BMJ 340: b5569, 2010) -- a growth factor in smoke? people who are strong enough / fit enough / hopeful enough / otherwise have more on-the-ball and can quit just do better? Be this as it may, the doctors are all over patients to get them to stop (Chest 143(5S): e61s-775, 2013).
PREINVASIVE LESIONS
* Autofluorescence bronchoscopy is now in use to find the squamous in-situ lesions. A majority will turn invasive (Chest 117: 1572, 2000).
CLINICAL MANIFESTATIONS OF BRONCHOGENIC CARCINOMA
The onset is notoriously subtle. Patients present with cough, chest pain, shortness of breath, and/or (especially) weight loss. The disease is most often unresectable when the patient comes to the physician.
Extension of the tumor in and near the chest causes many problems:
Metastatic disease:
It is very common for lung cancer to present as a brain tumor. Oat-cell is infamous for this, but any common type can do this.
{18764} brain "mets"
Bronchogenic carcinoma often involves the brain, bones, liver, adrenals (why?), kidneys, heart, pleura, and skin; no organ is immune.
{18774} adrenal "mets", this
may well have produced adrenal insufficiency
{18778} bone "mets", this hurts
Extrapulmonary, nonmetastatic manifestations of bronchogenic carcinoma are frequently seen. They include:
"Oat cell carcinoma causes autoantibodies against the nervous system because the cancer cells are of neural crest origin and the body is fighting them." This is probably mostly true.
| BRONCHIAL CARCINOIDS |

|
Most histologically low-grade lung carcinomas are typical carcinoids ("grade I neuroendocrine carcinomas") (Chest 119: 1647, 2001), from Kulchitsky cells. They stain with neuroendocrine markers.
The only known risk factor for these tumors is MEN-I. Tobacco smoking is not a risk factor.
Typical carcinoids are located in the large bronchi, exhibit a pushing border and are quite vascular (can hemorrhage after biopsy.)
Typical carcinoid by light microscopy (benign-appearing cuboidal cells in rows), immunoperoxidase (chromogranin-positive, neuron-specific-enolase positive, synaptophysin-positive), and electron microscopy (APUD granules).
Carcinoid syndrome is seldom produced by these lesions. Only about 10% metastasize, and even these tend to be indolent.
WHO-1999 distinguishes two other neuroendocrine (K-cell derived) lung cancers.
LARGE-CELL NEUROENDOCRINE CARCINOMAS (once called "intermediate oat cell", between carcinoid and oat-cell) are made of big cells with easy-to-see nucleoli, lots of mitotic figures, are neuron-specific-enolase positive but can be CD56-negative and/or chromogranin-negative and/or synaptophysin-negative, and have much larger cells than do classic oat-cell carcinomas. Almost all the patients are smokers. If they present at low stage, some are curable. Look for extensive necrosis, with cells palisading around it. It may not be worth distinguishing them from other large-cell carcinomas: Exp. Mol. Path. 70: 179 2001 -- and in fact, WHO-2004 reclassified it under large-cell undifferentiated.
OTHER TUMORS
* TUMORLETS ("chemodectomas"): harmless little meningioma-like things that are probably hyperplastic chemoreceptor cells. With today's imaging, we're finding these and they're turning up on the surgical pathology service. If the cytoplasm is scanty, they can mimic oat cell carcinoma. Leave the diagnosis to us.
![]() Carcinoid and oat cell
Carcinoid and oat cell
Lung pathology series
Dr. Warnock's Collection
* ALVEOLAR ADENOMA (today, probably "cystic mucinous carcinoma") is a very-slightly-malignant, multicystic tumor of older women. The cell of origin is the surfactant-producing pneumocyte.
* The misnamed "sclerosing hemangioma" is a benign tumor of type II pneumocytes, probably. It has many appearances but is always well-circumscribed. Leave the diagnosis to us.
LYMPHOMAS (including "lymphomatoid granulomatosis", a curious Epstein-Barr-related lymphoma with atypical lymphocytes and an intensive granulomatous response) are cancers, really-clonal (Am. J. Clin. Path. 103: 341, 1995). Today, we distinguish these from carcinomas using common lymphocyte antigen (lymphomas stain, carcinomas don't) and epithelial membrane antigen (carcinomas stain, lymphomas don't.) PSEUDOLYMPHOMAS (lymphoid hyperplasias -- "Arthur Godfrey's cancer") may turn into lymphomas in some cases.
* Ask a hemato-pathologist about LYMPHOMATOID PAPULOSIS.
PULMONARY HAMARTOMA: A growing nodule of cartilage, with clefts lined by pneumocytes. Future radiologists: recognize the familiar calcified, popcorn-shaped mass. If the radiologist can't call it, then it must be removed for fear of missing a cancer. Update on the benign masses that mimic lung cancer: Arch. Path. Lab. Med. 136: 1227, 2012.
|
|
![]() Lung hamartoma
Lung hamartoma
AFIP
Wikimedia Commons
{38506} lung hamartoma, gross
* SCLEROSING HEMANGIOMA / PNEUMOCYTOMA is a common benign tumor with epithelial and stromal cells both from the same neoplastic line -- leave the diagnosis to us.
* "Epithelioid hemangioendothelioma" is another low-grade malignancy tumor you can leave to us.
* PULMONARY BLASTOMA: A very rare, mixed epithelial and mesenchymal ("carcinosarcoma") cancer that resembles fetal lung. It occurs at any age, and there are no risk factors. It can be aggressive, but there are surgical cures.
* A host of "sarcomatoid" primary lung cancers are grouped separately by WHO-2004 and need not concern us.
* Occasionally a cancer with histopathology typical for salivary gland will pop up in lung instead -- again, these need not concern us.
METASTASES TO THE LUNG are common in many (if not most) carcinomas and sarcomas. Taking out lung metastases of sarcoma has long been popular, and nowadays the statistics are quite good for long-term survival (Clin. Orth. Rel. Res. 310: 188, 1995).
|
|
|
|
PULMONARY LYMPHATIC CARCINOMATOSIS (misnomer: "lymphangitic spread"): Seen in disseminated malignancy. Tumor plugs the pulmonary lymphatics, leading to pulmonary edema and rapid death.
{21001} pulmonary lymphatic carcinomatosis (white strings)
|
UPPER AIRWAY, LARYNX AND TRACHEA
|
|
NASOPHARYNGEAL CARCINOMA is fairly common. Around 25% are keratinizing squamous cell carcinomas, 15% non-keratinizing, and 60% undifferentiated (best prognosis, as radiosensitive). Remember the link between undifferentiated carcinoma and Epstein-Barr virus.
* SINONASAL ADENOCARCINOMAS are of two types. The "non-intestinal type" is seen in young folks and has an excellent prognosis. The "intestinal" type usually occurs in the ethmoid sinus and is more aggressive -- it's the one cancer for which formaldehyde gas might be a credible promoter, and wood dust and leather processing are also linked.
CONGENITAL ANOMALIES:
* The small larynx of people with del(5p) is responsible for the "cat cry."
SINUSITIS
Edema (allergy, infection) around the outlets of the sinuses sets up a vicious cycle, with non-drainage followed by bacterial infection followed by additional obstruction.
The most serious complication is infection of the cavernous sinus and nearby structures. Although sinus trouble is common, serious complications are rare.
|
|
INFECTIONS OF THE THROAT AND LARYNX are common:
THE COMMON COLD and VIRAL LARYNGITIS require no description.
EPIGLOTTITIS historically often resulted from infection with H. 'flu B. Airway obstruction is the major problem. It is terrifying for the child, who is reluctant to lie down because the epiglottis flops backward and causes obstruction. Deaths due from neglected infectious mononucleosis are usually from epiglottitis -- it responds quickly to glucocorticoids.
LARYNGOTRACHEOBRONCHITIS is more likely to be viral, particularly parainfluenza viruses; the taxonomy is given in NEJM 358: 384, 2008. Any can be called "croup" though classically this is a symptom with a barking ("like a trained seal") cough.
LARYNGEAL PAPILLOMATOSIS is a life-threatening infestation of the larynx with HPV (* usually strain 1).
![]() Laryngeal papilloma
Laryngeal papilloma
Lung pathology series
Dr. Warnock's Collection
TRAUMA most often results today from intubation (grisly article: Chest 96: 877, 1989). Forensic pathologists look for fractures of the hyoid bone in suspected strangling.
FOREIGN BODIES in the trachea can cause sudden death, especially in toddlers and drunken adults. I've seen a peach section, a roll of ham, and a denture, and a friend lost his two-year-old daughter to a peanut (keep these and popcorn away from toddlers). The "cafe coronary" is usually due to poorly-chewed beef.
LARYNGEAL NODULES ("Justin Timberlake's disease" -- dozens of famous vocalists have had these, though Mr. Timberlake's surgery got extensive coverage) are benign fibroepithelial polyps on the vocal cords of those who use their voices a lot -- drill sergeants, singers, teachers. They present as hoarseness and are easy to remove.
ANGIOFIBROMA OF THE NASOPHARYNX ("juvenile angiofibroma") is usually a teenaged boy's tumor (testosterone is a growth factor). This big white nuisance lesion is best left alone, since it bleeds copiously if biopsied.
NASOPHAGYNGEAL CARCINOMA, usually poorly-differentiated
squamous, is rampant in China and seems
to be caused by a combination of Epstein-Barr virus![]() (herpes 4)
(herpes 4)![]() and nitrosamine-loaded pickled fish
and pickled vegetables (update Br. J. Cancer 106: 206, 2012). How herpes 4
transforms cells: NEJM 333: 693 & 742, 1995.
and nitrosamine-loaded pickled fish
and pickled vegetables (update Br. J. Cancer 106: 206, 2012). How herpes 4
transforms cells: NEJM 333: 693 & 742, 1995.
* CANCER OF THE TRACHEA is rare and is usually adenoid cystic carcinoma or mucoepidermoid carcinoma, arising from tracheal glands that are homologous to salivary glands. (These can appear on the bronchi too.)
![]() Mucoepidermoid carcinoma and adenoid cystic carcinoma
Mucoepidermoid carcinoma and adenoid cystic carcinoma
Lung pathology series
Dr. Warnock's Collection
DYSPLASIA OF THE LARYNGEAL EPITHELIUM: Pre-cancerous, more or less aggressive depending on how mean it looks. Cancer 75: 457, 1995.
![]() Metaplasia in a smoker's larynx.
Metaplasia in a smoker's larynx.
Precedes dysplasia / cancer.
WebPath Photo
TB of the true vocal cords is transmissible by speaking, not just coughing.
CANCER OF THE LARYNX is very common, and is almost invariably a squamous cell carcinoma due to cigaret smoking. Ethanol abuse is also a risk factor; nobody knows why. HPV-16 is often present. There is usually some nearby carcinoma in situ.
Cancers confined to the true vocal cords (or the region just below them) are usually easy to cure using radiation, saving the voice. Cancers above the true vocal cords usually require laryngectomy.
{11696} laryngeal squamous cell carcinoma, supraglottic
|
|
* Subclassifying cancers of the larynx for wiser therapy: Ann. Ot. Rhin. Laryng. 104: 587, 1995.
PLEURAL DISEASES
"Devil's grip", or pleurodynia, is a complication of coxsackievirus A![]() or B
or B![]() infection in children (usually)
who are obviously sick with an acute febrile illness. It is frightening
but seldom dangerous.
infection in children (usually)
who are obviously sick with an acute febrile illness. It is frightening
but seldom dangerous.
PLEURAL EFFUSION -- fluid in the pleural space -- is a ubiquitous problem in clinical medicine. (Future radiologists: Before you can see it on x-ray, there must be about 700 mL -- a lot of fluid).
Each side can actually hold about 4000 mL if the lung is completely collapsed. Effusions cause atelectasis and restrict lung movement, you can tap them for diagnostic or therapeutic purposes, you can prevent their buildup by sclerosing the pleural space, etc.
TRANSUDATES: congestive heart failure, nephrotic syndrome, cirrhosis. All other pleural effusions will probably be exudates.
HYDROTHORAX: pleural effusion that is a transudate or serous exudate.
HEMOTHORAX: a pleural effusion that is blood
![]() Hemothorax
Hemothorax
Urbana Atlas of Pathology
PYOTHORAX or EMPYEMA: a pleural effusion that is pus
CHYLOTHORAX, from injury to the thoracic duct
PNEUMOTHORAX: Air in the pleural space, i.e., the lung has collapsed.
This can result from a wound to the chest wall (why?), or from a tear in the visceral pleura. The latter can happen in healthy people ("spontaneous pneumothorax") or from tumor or injury (iatrogenic, pink puffer).
* Catamenial pneumothorax accompanies menstruation, usually in multiparous women. The remedy is to do pleurodesis with talc during the menstrual period.
Pneumothorax is most serious ("tension pneumothorax") if a valve-like flap on the visceral pleura lets air into the chest wall but not out.
"Three Kings". George Clooney treats teammate... |

...Mark Wahlberg's tension pneumothorax |
PLEURAL PLAQUES of dense collagen are harmless but are a marker for asbestos exposure. They have nothing to do with smoking.
BENIGN MESOTHELIOMA is a pedunculated nubbin made of fibrous tissue. It has nothing to do with asbestos.
MALIGNANT MESOTHELIOMA ("Steve McQueen's disease"): cancer of the pleural mesothelium. Update Arch. Path. Lab. Med. 136: 882, 2012.
This always-incurable cancer usually results from asbestos exposure, which can be documented in about 80% of cases.
* Turkey has some asbestos-rich soils and in these regions, blown dust may explain the abundance of asbestos fibers in the lungs and the high prevalence of mesotheliomas. See Env. Health Perspect. 108: 1047, 2000; Chest 143: 164, 2013. In asbestos-exposure-negative mesothelioma (Arch. Path. Lab. Med. 136: 262, 2012), exposure to the nonasbestos fiber mineral erionite, therapeutic radiation, perhaps SV40 virus exposure are the risk factors; there's even talk of carbon nanotubules.
The usual picture is that of a biphasic tumor (why?); histologic subtyping into "epithelioid" and "sarcomatoid" variants is of little use in prognosticating survival (Thorax 44: 496, 1989). Distinguishing mesothelioma from look-alike cancers and from benign mesothelial hyperplasia is difficult. Tips for future pathologists:
Negative in mesothelioma: CEA (carcinoembryonic antigen), TTF-1 (famous lung cancer marker)
As a matter of fact, telling epithelial mesothelioma from reactive hyperplasia of the mesothelium can be difficult, in biopsies or effusions. Telling a desmoplastic spindle cell mesothelioma from a pleural scar is equally treacherous. Clear-cut stromal invasion is the only reliable way of being sure it is cancer.
Pathologists can now tell it from metastatic disease most of the time by staining for keratins of various MW's -- a wide range are present in mesothelioma. Autopsy is still helpful in confirming the diagnosis.
* And this may all be moot if fibulin-3 turns out to be a sensitive and specific serum marker for pleural mesothelioma (NEJM 367: 1417, 2012).
|
|
|
|

|
|
|
|
Oddly, a few percent of mesotheliomas produce insulin-like molecules that cause hypoglycemia.
* An ultra-inbred community in Turkey in which 50% of the villagers die of mesothelioma, with only a dubious link to environmental carcinogens: Lancet 357: 444, 2001.
Rule: The parietal pleura is very sensitive to pain, far more so than the lung or any but the largest airways. If chest pain accompanies respiratory movements, the cause is probably inflammation or irritation of the parietal pleura. You may have experienced the "stitch", caused by a wrinkle and relieved by super-inflating your lungs.
|
|
|
{27597} mesothelioma, gross
* The Fellowship of those who bear the Mark of Pain. Who are the members of this fellowship? Those who have learned by experience what physical pain and bodily anguish mean, belong together all the world over; they are united by a secret bond. Praise God! One and all they know the horrors of suffering to which man can be exposed, and one and all they know the longing to be free from pain. He who has been delivered from pain must not think he is now free again, and at liberty to take life up just as it was before, entirely forgetful of the past. He is now a "man whose eyes are open" with regard to pain and anguish, and he must help to overcome those two enemies (so far as human power can control them) and to bring to others the deliverance which he has himself enjoyed. The man who, with a doctor's help, has been pulled through a severe illness, must aid in providing a helper such as he had himself, for those who otherwise could not have one. He who has been saved by an operation from death or torturing pain, must do his part to make it possible for the kindly anaesthetic and the helpful knife to begin their work, where death and torturing pain still rule unhindered. The mother who owes to medical aid that the child still belongs to her, and not to the cold earth, must help, so that the poor mother who has never seen a doctor may be spared what she has been spared. Where a man's death agony might have been terrible, but could fortunately be made tolerable by a doctor's skill, those who stood around his deathbed must help, that others, too, may enjoy that same consolation when they lose their dear ones.
Such is the Fellowship of those who bear the Mark of Pain.
-- Albert Schweitzer, M.D., Ph.D.
On the Edge of the Primeval Forest
* SLICE OF LIFE REVIEW
{11512} lung, normal
{11739} lung, normal
{14900} epiglottis, normal
{14901} olfactory epithelium, normal
{14902} olfactory epithelium, normal
{14903} trachea (false & true vocal cords)
{14904} trachea (false & true vocal cords)
{14905} trachea, normal
{14906} respiratory epithelium (of trachea)
{14907} respiratory epithelium (of trachea)
{14908} bronchus, normal
{14909} bronchus, normal
{14910} bronchiole, normal lung
{14911} bronchiole, normal lung
{14912} respiratory epithelium, normal
{14913} respiratory bronchiole, normal
{14914} respiratory bronchiole, normal
{14915} alveolar duct, normal
{14916} alveolar duct, normal
{14917} alveolus, normal
{14918} alveolus, normal
{15145} trachea
{15147} trachea
{15148} lung
{15149} lung
{15150} lung, normal
{15151} lung, normal
{15152} lung
{15154} pneumocytes, type Ii lung
{15289} trachea, normal
{15290} trachea, normal
{15291} epiglottis, normal
{15292} epiglottis, normal
{15293} epiglottis, normal
{15294} trachea, * seromucous glands
{15295} trachea, normal
{15297} lung, normal
{15298} bronchiole, respiratory bronchiole
{15299} pneumocyte, type Ii
{15300} bronchus, normal
{15302} bronchiole, normal
{15303} bronchus, * seromucous gland
{15763} lung, normal
{15764} lung, normal
{15765} lung, normal
{15766} lung, normal
{15767} lung, normal
{15768} lung, normal
{15791} larynx, normal
{15792} larynx, normal
{17521} bronchus, normal
{17566} lung, normal
{20898} trachea
{20899} trachea, seromucous gland
{20900} cilia, tracheal epithelial cells
{20902} epiglottis
{20903} epiglottis
{20904} trachea
{20907} trachea, epithelium
{20909} alveolus, lung
{20910} lung, respiratory duct
{20911} lung, terminal bronchiole ?
{20912} lung, terminal bronchiole ?
{20913} bronchus, lung
{25654} lung, normal
{27215} lung, normal
{27218} lung, normal
{27221} lung, normal
{27224} lung, normal
{27227} lung, normal
{27468} lung, normal
{29168} bronchitis, chronic with normal to compare
{37604} lung, normal
{37619} bronchus, normal
{41517} lung, normal cast of capillary bed
{46452} xerogram laryngeal, normal
{15289} trachea, normal
NOTE: You may not share Mr. Housmann's "scientific" nihilism. I'm always surprised and pleased when such folks at least pay lip service to basic human goodness.
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about lung pathology to consult these standard textbooks.
In my notes, the most helpful current journal references are embedded in the text. Students using these during lecture strongly prefer this. And because the site is constantly being updated, numbered endnotes would be unmanageable. What's available online, and for whom, is always changing. Most public libraries will be happy to help you get an article that you need. Good luck on your own searches, and again, if there is any way in which I can help you, please contact me at scalpel_blade@yahoo.com. No texting or chat messages, please. Ordinary e-mails are welcome. Health and friendship!


| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |


 Pulmonary Pathology
Pulmonary Pathology

