Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
verify here. |
Cyberfriends: The help you're looking for is probably here.
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
QUIZBANK
Blood & Lymph #'s 133-139, 178-333
 |
Describe the distribution of lymphoid tissue in humans, with special reference to B- and T-cell zones. Describe the microanatomy of the lymph nodes. Sketch the sequence by which a B-cell develops into a plasma cell, and name each stage. Distinguish relative and absolute counts of various white cells, and explain why absolute counts are more meaningful. Calculate an absolute count by multiplying the total and percentage counts. Give the healthy absolute counts for lymphocytes, monocytes, eosinophils, and neutrophils. Given a name of a white cell marker, tell what cell(s) it identifies. Given a white cell type, mention its major markers. Given a patient with neutropenia and a history, come up with a reasonable differential diagnosis. Describe the typical cause and course of agranulocytosis. Recognize the major causes of lymphopenia. Give a reasonable differential diagnosis for granulocytosis, eosinophilia, and lymphocytosis. Tell how to distinguish chronic myelogenous leukemia from leukemoid reaction. Describe possible peripheral (i.e., circulating) white cell pictures in sepsis. Mention the significant disease association for increased absolute basophil count. Describe the important non-neoplastic ("reactive") causes of lymphadenopathy, and how each looks under the microscope. Describe "infectious mononucleosis syndrome", and name its four principal etiologic agents. Explain how a pathologist distinguishes a malignant lymphoma from a worrisome reactive (benign) lymph node. Do this yourself for an easy case. Apply the unifying "rules" in this handout to clinical problems about non-Hodgkin's lymphomas. Explain how the classic Rappaport system differs from the International Working system and the Revised European-American system of lymphoma nomenclature. Recognize the names of the low, middle, and high grade lymphomas. Given the name of a non-Hodgkin's lymphoma, recognize its distinctive features. Identify non-Hodgkin's lymphomas based on their idiosyncratic markers, etiologies, or epidemiologies. Explain current thinking about the pathogenesis of Hodgkin's disease. Describe its epidemiology, subtypes, and prognosis. Given a description of the background, name the subtype, and vice versa. Give a short account of the World Health Organization's system of classifying the acute leukemias, and directions that future classifications will probably take. Describe the major kinds of leukemia in detail. Cite their etiologies (if known), pathogenesis, natural histories, subclasses, diagnostic features, and current prognosis. Do the same for the myelodysplastic syndromes, polycythemia vera, and "myelofibrosis with myeloid metaplasia". Describe the pathogenesis, symptoms, signs, lab findings, diagnosis, typical course, and major complications of plasma cell myeloma. Recognize and prognosticate the other "plasma cell disorders". Recognize the noteworthy causes of polyclonal gammopathy. Explain current thinking about Langerhans cell histiocytosis (histiocytosis X). Given an enlarged spleen and the opportunity to ask questions, come up with a reasonable differential diagnosis. Describe the common findings in spleens at autopsy. Name the lymphoma and/or leukemia caused by with each of these viruses: Epstein-Barr virus Correctly define and use the following terms: agranulocytosis
Identify the following elements in peripheral and/or marrow smears: all five type of normal white cells
Shown an appropriate peripheral smear, tell when each disease might be present: Pelger-Huet anomaly
Identify all the following cells in microscopic sections: normal lymphocytes
Identify each of the following disease patterns under the microscope: follicular hyperplasia
Draw or recognize a Birbeck granule and describe its significance. |
INTRODUCTION
You will refer to this material every time you feel a large lymph node or spleen, or have a patient with an abnormal CBC.
"Leukemias and lymphomas" is the most difficult unit in Medical Pathology except for glomerular disease. You can't learn it if you are not continually asking yourself, "Why?"
You are already familiar with the development of the different kinds of white cells, and the locations of lymphoid tissue throughout the body (lymph nodes, Waldeyer's ring, Peyer's patches, spleen, large airways).
T-cell zones: thymus, lymph node parafollicular cortex, splenic white pulp near arteriole
B-cell zones: germinal centers and their mantles, splenic white pulp at its margins
Among circulating lymphocytes, 80% are T-cells, and 20% are B-cells.
* You are also familiar with the common reaction patterns of various white blood cells: acute inflammation, pus, granulomas, and accumulations within phagocytes. (There's no need, for example, to talk right now about xanthomas, lipogranulomas, etc., etc.)
In discussing diseases that affect numbers of white blood cells in the peripheral blood, it is much more useful to talk about ABSOLUTE CELL COUNTS than "percentage counts".
Of course, you can estimate the absolute count by multiplying the total WBC count x the % for a particular cell.
Healthy absolute counts:
Basophils: * few- 100/cu mcL (heads up -- basophil granules are soluble and can wash out during slide preparation)
Eosinophils: few- maybe 400 (fewer in AM, more in PM)
Lymphocytes: 1200-3400 (* 3000-7000 for kids)
T4 helper lymphocytes: >=1000
Monocytes: 100- 590
Neutrophils: 1800-6500
Note that "95% lymphocytes" might mean either agranulocytosis (if the total white count is 2000) or chronic lymphocytic leukemia (if the total white count is 100,000). This is why I like white cell differential counts reported in absolute numbers, and why all labs do this nowadays.
* Current smokers average 25% higher neutrophil counts; those who've quit in the last five years still average higher (Am. J. Clin. Path. 107: 64, 1997). This won't matter in your clinical decision-making.
A good "normal range" for total white count is 4000-11000/cu mcL. "Leukocytosis" is present when the white count exceeds 12,000/cu mcL.
"Reactive" (infection, immune, meds) proliferations of white cells produce big lymph nodes / spleens and/or high counts of various blood cells. They are ubiquitous and usually trivial, secondary to whatever-else if a cause is even found. You probably have 2-5 palpable lymph nodes in your neck. They are your friends. The really important "white cell diseases" are neoplastic. These are:
(1) the MALIGNANT LYMPHOMAS (HODGKIN'S AND NON-HODGKIN'S), solid tumors of lymphocytes (the rare tumors that truly arise from monocyte-macrophages are also included here; no one knows the true cell of origin of the malignant cells of Hodgkin's disease, which is also included here)
(2) the LEUKEMIAS and their close relatives, the MYELOPROLIFERATIVE DISORDERS, in which sick hematopoietic stem cells proliferate
(3) the PLASMA CELL DISORDERS, which typically produce antibodies and/or fragments thereof
(4) the LANGERHANS CELL HISTIOCYTOSIS FAMILY ("histiocytosis X"; "disseminated histiocytosis") of quasi-cancers, much less common than the others
Probably because it is so easy to harvest the cells, and since chemotherapy has been more successful for these diseases than for most other cancers, a tremendous amount of study has gone into clarifying their molecular pathology.
There can be no such thing as a truly benign neoplasm of white blood cells, since by their very nature they infiltrate tissues. Some of these entities (for example, the acute leukemias) are far more aggressive than others ("benign plasmacytoma", "benign monoclonal gammopathy").
White cell markers that an ordinary physician might actually want to know:
TdT: immature lymphocytes
E-rosettes: T-cells
{16282} E-rosette, around a T-cell
CD3, CD4, CD8, αTcR, βTcR, γTcR, δTCR, others: the more mature T-cells (various kinds)
CD1a (T6): some T-cells, all Langerhans macrophages
CD5: mantle cell lymphoma, many CLL (depends on the mutation)
* CD10 (CALLA): most B-cells (mantle cell lymphoma is negative)
CD15: Most Reed-Sternberg cells; some others
* CD19: B-cells, but not plasma cells
* CD20: all but the most primitive B-cells, but not plasma cells
* CD22: most B-cells (EBV receptor)
* CD34 : primitive blood cells -- great for counting "blasts" in leukemia / preleukemia
CD45 ("common leukocyte antigen" / LCA): all white cells (* exception: Reed Sternberg cells and some leukemias)
* CD65: Most consistent marker for the natural-killer lymphocytes, non-B, non-T cells making up maybe 10% percent of your circulating white cells. (They tend to be big and have granules. Update on their neoplasms: Cancer 112: 1425, 2008).
CD68: common macrophages
* CD79a: The mantle lights up best
BCL-2 (apoptosis-preventer): turned OFF during hypermutation (i.e., germinal centers); turned ON in most nodular / follicular lymphomas
surface Ig(M, etc): B-cells
kappa, lambda: mature B-cells, plasma cells -- especially useful for showing clonality (i.e., neoplasia)
cyclin D1: mantle cell lymphoma stains strongly
cytoplasmic Ig: plasma cells
* nonspecific esterase: monocytes
Fc receptor: B-cells, monocytes
TRAP: hairy-cell leukemia
HLA-D/DR /Ia: Langerhans cells and other antigen-presenting macrophages; some other cells
lysozyme: monocytes
* alpha1-antichymotrypsin: monocytes
erythrophagocytosis: monocytes
(myelo-)peroxidase: granulocytes
* Sudan black: granulocytes
* chloroacetate esterase: neutrophils, basophils, mast cells
platelet markers: megakaryocytes
* PAS+ diffusely: erythrocytes, megakaryocytes/platelets
* PAS+ chunks ("blocks"): immature lymphocytes or M6 leukemia
S-100, CD1/T6: dendritic ("Langerhans") macrophages
I would ask you NOT to worry about differentiation markers beyond what's been listed above. A pathologist MUST know them, as a Hodgkin or non-Hodgkin lymphoma cannot be properly classified without immunohistochemistry (update Arch. Path. Lab. Med. 132: 441, 2008). A sub-subclassification for epidemiologists: Blood 110: 685, 2007.
* {16517} neutrophil, chloroacetate esterase stain
![]() Neutrophilia
Neutrophilia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
NEUTROPENIA: A low absolute neutrophil count in the peripheral blood for any reason. An absolute neutrohil count below 100 is life-threatening. A count below 500 is considered "severe"; 500-1000 is "moderate", 1000-1500 is "mile". (NOTE: "Leukopenia" is a not-very-useful word that describes any low total white count. Since most granulocytes are neutrophils, we can also call neutropenia "granulocytopenia")
Possible causes include
SUPPRESSION OF GRANULOPOIESIS
"The aplastic anemias" (better, "bone marrow failure")
Bad stuff in the marrow
Space-occupying lesions ("myelophthisic anemias")
Solid cancers
Granulomas
Hematologic malignancies that suppress granulopoiesis (i.e., some leukemias and lymphomas)
DNA problems
Cancer chemotherapy
Radiation sickness
"The megaloblastic anemias"
Hereditary cyclic (q. 3 wk., severe; dominant mutation usually in the ELA2 elastase gene, molecular biology Blood 92: 2629, 1998; mutated neutrophil elastase that itself damages the cellular machinery; also Nat. Genet. 35: 90, 2003; Blood 108: 493, 2006; there is an even worse variant "severe congenital neutropenia")
* Shwachman-Diamond (genetic, also fatty pancreas; "cystic fibrosis" mimic with normal sweat chloride test)
Typhoid fever
Occasional virus infections (mild suppression, especially parvo B19![]() (Am. Fam. Phys. 75: 373, 2007))
(Am. Fam. Phys. 75: 373, 2007))
* Some childhood acute leukemias going back to the stem cells
* LGL leukemia -- these T-cells suppress young granulocytes
* MYELOKATHEXIS (group of genetic diseases with accelerated neutrophil precursor apoptosis; surviving neutrophils are hypersegmented and have very long bars between nuclear lobes: Blood 95: 320, 2000; Am. J. Hem. 62: 106, 1999).
* Kostmann's -- genetic disease (several loci); almost all of the developing neutrophils die at the myelocyte stage. The usual cause of very low absolute neutrophil count at birth and after.
Idiopathic
* The lab machine didn't count them.
EXCESS DESTRUCTION / SEQUESTRATION OF NEUTROPHILS
Autoimmune (rare, think of lupus)
Hypersplenism (see below)
Sequestration in a rapidly-growing abscess (??)
Idiopathic
DRUGS: The mechanisms are typically obscure (Ann. Int. Med. 146: 657, 2007).
The most common offenders today:
PERSONAL-TRIVIAL
Some people of African descent people just have slightly low neutrophil counts
Some women get a mild neutropenia around their periods
AGRANULOCYTOSIS is a time-honored misnomer for neutropenia sufficiently severe to put a person at risk for serious infection (i.e., neutrophil counts of 1000 or less, often much less; <500 is really an emergency).
The first sign is typically mouth ulcers ("there's lots of germs in there") with their pseudomembranes laden with infectious bacteria and/or fungi.
Later, the body is overwhelmed by bacteria, with death ensuing in a few days. Until the very end, patients are likely to complain only of "just not feeling quite right".
The usual cause of "agranulocytosis" problems is medications. Future docs: If you notice that somebody has an absolute neutrophil count <1800 or so but not in the danger zone, stop all medications that can be stopped, and check again in a week.
LYMPHOCYTOPENIA is less common and less perplexing than neutropenia. Think of hereditary immunodeficiency, HIV, radiation injury, marasmus/kwashiorkor, Cushing's syndrome, or just "stress".
In systemic viral infections, interferons may drive helper T-cells into the lymph nodes and/or onto receptors on endothelial cells. No big deal.
LEUKOCYTOSIS: It's worth remembering the following NON-NEOPLASTIC CAUSES OF ELEVATED WHITE CELL COUNTS. Most of them make sense:
You remember that in health, about half the neutrophils in the blood are circulating, and the other half are marginated on vessel walls ("the marginal pool"), at any time.
LOTS OF NEUTROPHILS ("granulocytosis"):
(don't forget surgery and myocardial infarcts)
This explains the "pop" claim that "exercise greatly enhances immunity" (fallacy showcased JAOA 96: 166, 1996). I am also not aware of any reason whatever to believe in the claimed ""open window to infection" following the third hour of endurance events.
Glucocorticoids also prevent neutrophils from entering tissues (among other things, they down-regulated selectin on their surfaces)
Lithium for some reason tends to increase neutrophil production, often doubling it
NOTE: Typhoid patients and some super-septic patients may become neutropenic because granulopoiesis is suppressed and/or all the neutrophils have emigrated from the blood. Beware of relying on white count as your chief marker for infection!
NOTE: The super-sick, septic patient is likely to have TOXIC GRANULATION (extra-prominent azurophilic granules), CYTOPLASMIC VACUOLES ("from doing all that phagocytosis"), and/or DOHLE BODIES (rough endoplasmic reticulum remnants). By contrast, if the neutrophil count simply rises from acute pain and "stress", there will be no toxic granulation, vacuolization, or left shift. More about these in "Clinical Pathology".
{13646} Dohle body
{13661} Dohle body
{16213} Dohle body
* Future pathologists: The latter two "Dohle bodies" are fakes; they are from cases of May-Hegglin's (say "Muh-HAY-lun") semi-disease, an autosomal dominant trait (MHY9) with too-few, too-big platelets and lots of "Dohle bodies" and big granules; the neutrophils function normally. May-Hegglin "Dohle bodies" are actually non-muscle myosin A, gene mutated in May-Hegglin: Nat. Genet. 26: 106, 2000; Blood 97: 1147, 2001. There are several different phenotypes at the locus (Blood 102: 529, 2003).
NOTE: LEFT SHIFT refers to presence of immature white cells ("bands") in the peripheral blood, i.e., they're being
mobilized early from the bone marrow. To tell an extreme case (WBC's up to 100,000 or so, i.e., a
LEUKEMOID REACTION, as in sepsis, overwhelming TB![]() , or carcinomatosis) from chronic granulocytic
leukemia (see below), remember the following:
, or carcinomatosis) from chronic granulocytic
leukemia (see below), remember the following:
(1) In chronic granulocytic leukemia, the LEUKOCYTE ALKALINE PHOSPHATASE tends to be low. In sepsis and the non-leukemic myeloproliferative disorders, it tends to be high.
Leukocyte alkaline phosphatase is a completely different test from the "serum alkaline phosphatase" on the chemical profile. DON'T talk about them together.
(2) In chronic granulocytic leukemia, the ABSOLUTE BASOPHIL COUNT is generally high, too. This would be unusual in sepsis.
(3) In chronic granulocytic leukemia, there is virtually always a switch of material between chromosomes 9 and 22 (i.e., the PHILADELPHIA CHROMOSOME (Ph') or at least its molecular equivalent). You won't see this except in cancer.
(4) And of course, toxic granulation (very easy-to-see granules on stained blood; nobody really why)/ toxic vacuolization says "infection", not "leukemia".
(5) When in doubt, it's a leukemoid reaction. An indolent leukemia can wait for a few days; deadly infection can't.
Philologists: RIGHT SHIFT refers to the hypersegmented granulocyte nuclei of pernicious anemia (etc., any major impediment to normal DNA synthesis will produce this "megaloblastic" change). "Right" and "left" derive from spaces on the old do-it-by-hand tally sheets.
LOTS OF EOSINOPHILS (big review Mayo Clin. Proc. 80: 75, 2005; bigger review Arch. Path. Lab. Med. 137: 259, 2013):
The "Loeffler" family of eosinophil-mediated diseases -- now being sorted out; while most remain idiopathic, a few have known mutations
CHRONIC EOSINOPHILIC LEUKEMIA, an entity removed from the Loeffler's wastebasket by the discovery of FIP1L1-PDGFRalpha (Haematol 95: 696, 2010) -- treat with imatinib.
Type I immune injury
food allergy, hay fever, eczema, extrinsic asthma (supposedly -- you won't be impressed)
bronchocentric granulomatosis (aspergillus superinfection in asthma; this one's important)
* hyper-IgE ("Job's") immunodeficiency
Tissue parasites
ascariasis
filariasis (includes "tropical eosinophilia" of the Far East -- future pathologists: filaria worms will be pushed to the "feather edge" of the smear)
onchocerciasis
strongyloidiasis
trichinosis
echinococcus
schisotomes / katayama fever
visceral larva migrans (dog and cat roundworms)
cutaneous larva migrans (dog and cat hookworms)
Drug allergy (most any; but notoriously gold therapy for arthritis, where eosinophilia is almost expected)
Hodgkin's disease (a large minority of cases)
Churg-Strauss (a vasculitis, often with granulomas, usually with ANCA; it's not clear whether this is a separate disease, or simply the way Wegener's / polyarteritis manifests in folks with allergies)
Dermatitis herpetiformis
Familial hypereosinophilia (locus unknown, autosomal dominant, mild: Blood 103: 4050, 2004)
* Well's eosinophilic cellulitis
Eosinophilia-myalgia syndrome (from the tainted tryptophan)
* Any AIDS patient with a rash (Am. J. Med. 102: 449, 1997)
* Pemphigus (I don't know why)
* Dermatitis herpetiformis
* Crohn's
* Transiently during atheroembolization
* Acute liver transplant rejection (almost all have it, no one knows why)
Dermatomyositis
Polyarteritis nodosa (don't miss this one)
* Kimura's angiolymphoid hyperplasia with eosinophilia (very high IgE, eosinophil-lymphoid pseudotumors of head and neck, germinal centers loaded with eosinophils; marked peripheral eosinophilia; common in middle-aged Asian men, Asia, rare elsewhere; making the call Pediatrics 110: e-39, 2002; probably a low-grade lymphoproliferative disorder Am. J. Surg. Path. 26: 1083, 2002; Arch. Path. Lab. Med. 131: 650, 2007)
* mastocytosis with eosinophilia (molecular signature known, response to imatinib/Gleevic likely)
* T-cell neoplasms making interleukin-5: NEJM 341: 1141, 1999
* "Clonal eosinophilia" -- FIP1L1-PDGFRA fusion gene
NOTE: In the developed world, among clinically healthy patients with isolated elevated eosinophil counts, you will often not find the cause.
NOTE: I did CBC's for years on medical students, many of whom have hay fever, etc., and have never found one with an elevated eosinophil count.
NOTE: Remember that eosinophilic counts are up in the afternoon and down in the morning because the morning's cortisol surge suppresses them; I'd suggest taking a serious look at an absolute eosinophil count over 350 or so in the morning, and over 650 in the afternoon.
NOTE: The "Loeffler's eosinophilic" problems are a curious, mixed-bag of diseases with excessive numbers of eosinophils in various tissues that cause tissue damage.
Sometimes the underlying problem is a proliferation of mutated T-cells producing excessive eosinophil attractants (NEJM 330: 35, 1994).
* In other cases, the eosinophils themselves seem to be the mutated clone: Blood 93: 1651, 1999.
In 2004, I predicted the success of the anti-IL5 antibody mepolizumab as treatment (J. Allerg. Clin. Imm. 113: 115, 2004); it has been a spectacular success (NEJM 358: 1215, 2008).
{14099} eosinophilic leukocytes (buffy coat)
{09207} eosinophil granule with crystal (electron micrographs; these crystals will combine to form large
Charcot-Leyden crystals under some conditions)
![]() Eosinophilia
Eosinophilia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
LOTS OF MONOCYTES -- Chronic stuff, seldom helpful, and it NEVER makes the diagnosis:
typhoid fever
bad granulomatous problems
TB
brucellosis
Crohn's disease
leprosy![]()
deep fungi
sarcoidosis
others
* chronic autoimmune disease
rheumatoid arthritis is worth remembering
* rickettsial disease (often; red flag)
* disseminated cancer (occasionally)
LOTS OF LYMPHOCYTES:
"infectious mononucleosis" (see below)
whooping cough ("pertussis"; little cleaved lymphocytes; the toxin keeps the T-cells from homing to lymphoid tissue; Am. J. Clin. Path. 114: 35, 2000)
infectious lymphocytosis (mild kids' disease, with T-cells,
caused by various non-herpes viruses notably coxsackie B2![]() ; a "chronic form" also exists without
marrow abnormalities; leave this to the pediatric hematologists; Acta. Paed. 74: 633, 2008.)
; a "chronic form" also exists without
marrow abnormalities; leave this to the pediatric hematologists; Acta. Paed. 74: 633, 2008.)
"transient stress lymphocytosis" (absolute counts 4000-10000; on the evidence we've overlooked this for years; all major lymphocyte subsets go up, and neutrophils go up too: Am. J. Clin. Path. 117: 819, 2002)
* really bad "collagen-vascular disease"
* phenytoin ("Dilantin") or para-amino salicylic acid ("PAS") therapy
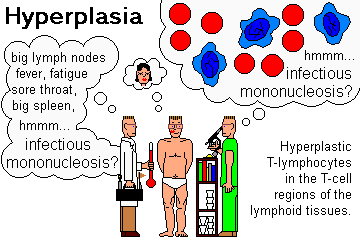
NOTE: INFECTIOUS MONONUCLEOSIS is a family of diseases featuring fever, malaise, fatigue,
lymphadenopathy, and circulating benign atypical lymphocytes. The syndrome results from first
meeting one of these four micro-organisms: (1) Epstein Barr virus![]() ;
(2) cytomegalovirus
;
(2) cytomegalovirus![]() ;
(3) toxoplasmosis
;
(3) toxoplasmosis![]() ;
(4) HIV.
;
(4) HIV.
BENIGN ATYPICAL LYMPHOCYTES are activated cells (B- or T-) seen typically in the blood in "infectious mononucleosis" and certain other infections; you may see a few in any viral illness.
Probably more important in spotting infectious mono and telling it from leukemia when you're a beginner is that when there are bunch of "atypical lymphocytes", no two look the same.
|
|
LOTS OF BASOPHILS:
chronic myelogenous leukemia
other "chronic myeloproliferative disorders"
polycythemia vera
* primary hemorrhagic ("essential") thrombocythemia
* supposedly up a little bit in lots of other things; this will not be important clinically.
NOTE: None of these "classic findings" is either particularly sensitive, or particularly specific, for any particular disease. Use this information in the setting of the "whole person".
ODD NEUTROPHILS:

We have already mentioned CHRONIC GRANULOMATOUS DISEASE, a poorly-named group of defects in the ability of neutrophils to kill common bacteria, with the macrophages needing to become involved as well.
|
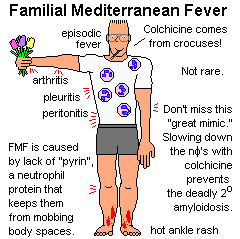
FAMILIAL MEDITERRANEAN FEVER, long-mysterious, has now
yielded up its secrets.
Lacking pyrin, neutrophils mob body cavities every once in a while.
In addition to fever, patients may have pleuritis, arthritis,
peritonitis, and/or a hot rash (looks like a strep Colchicine, famous for its ability to slow down neutrophils (as in acute gout), controls the attacks and prevents the dread complication of secondary amyloidosis. IL-1-related new biologicals help people who seem refractory (Clin. Pharm. Ther. 95: 89, 2014.) As you can imagine, acute FMF can mimic many diseases. The amyloidosis AA that often develops in these patients can mimic most of the rest. Don't miss it.
* A similar, thankfully-rare periodic fever syndrome is caused by a mutation in the TNF-receptor (TNFR1): Blood 108: 1320, 2006. Yet another is caused by mutated cryopyrin (includes serious neurologic problems; anti-interleukin-1 treatment brings about full resolution: Neurology 74: 1267, 2010). |
|
You recall CHEDIAK-HIGASHI SYNDROME, in which there are several problems with organelle membrane synthesis. synthesis.
Melanosomes don't form properly, so there is partial albinism.
The lack of platelet dense granules results in a bleeding tendency.
* The gene has been cloned (LYST, lysosomal traffic regulator). Most of these patients go on to develop a lethal non-neoplastic hyperplasia of the lymphocytes. Marrow / stem cell transplant is now routine and prevents this. Review Blood 95: 979, 2000.
* There is a report that long-term survivors of bone marrow transplantation develop a neurodegenerative disease after decades (Blood 106: 40, 2005). Stay tuned.
|
In the autosomal dominant PELGER-HUET ANOMALY, the neutrophil nuclei fail to segment normally, producing
"peanuts" and "pince-nez eyeglasses". ("Look! This clinically healthy patient has a horrible left shift / leukemia!")
This is a fairly common laboratory curiosity (maybe one person in 5000), and is usually of no significance.
The gene is LBR, involved in keeping the nucleus's shape; there are a few troublesome
alleles but the common ones are not a problem even for heterozygotes.
Explain to the physician and the patient, and check their close kin before somebody gets sick
and everybody gets confused (Am. J. Clin. Path. 137: 358, 2012). (* Double doses get no
segmentation whatever. And they don't seem to have any obvious troubles
with bacteria or
anything else: Acta. Hem. 66: 59, 1981. "Look! This clinically healthy patient has all myelocytes!"
"No, look around, they're pelgeroid.")
* Acquired / pseudo-Pelger-Huet can been seen when there are mutations (i.e., myelodysplasia, leukemia) or for some mysterious reason as a medication side-effect (Arch. Path. Lab. Med. 130: 93, 2006; Am. J. Clin. Path. 135: 291, 2011). These tend to be a minority of neutrophils, tend to be hypogranular, the lobes aren't equal-sized as in Pelger-Huet, and they tend to have denser chromatin -- perhaps they are dying. Let us worry about them. | 
|
{16208} Pelger-Huet, one dose
{16209} Pelger-Huet, one dose
{13658} Pelger-Huet, two doses
* ALDER-REILLY ANOMALY merely refers to large, mucopolysaccharide-laden granules in some of the storage diseases (Hunter's, Hurler's, Tay-Sach's, occasionally as an acquired trait in myelodysplasia). You will see it in all five types of white blood cells. Don't mistake this for "toxic granulation."
* Thankfully rare: Lack of endothelial adhesion molecules for phagocytes (J. Clin. Invest. 103: 97, 1999) or lack of CD18 integrin on neutrophils (Blood 91: 1520, 1998).
Bacilli in neutrophil vacuoles: Usually Capnocytophagia canimorsus (DF2, dog bite)
* And you know that drumsticks are the inactivated X-chromosomes of lyonization.
MORULAE OF EHRLICHIOSIS![]() can help you diagnose
this famous "spotless fever"; this "granulocytic" variant
of ehrlichiosis can be fatal (NEJM 334: 209, 1996). "Morule" is Latin for "mulberry".
can help you diagnose
this famous "spotless fever"; this "granulocytic" variant
of ehrlichiosis can be fatal (NEJM 334: 209, 1996). "Morule" is Latin for "mulberry".
NORMAL LYMPH NODE ANATOMY
LYMPH NODES are soft (i.e., reticulin-framework) ovoids, up to about 2 cm in health. Afferent lymphatics penetrate and travel within their capsules (metastatic cancer first sets up here). Afterwards, lymph percolates through the cortex, and then the medulla, leaving by the hilum.
Within the cortex, there are generally some germinal centers ("lymphoid follicles"), sites of actively-proliferating B-cells. Each germinal center is surrounded by a mantle of resting B-cells, which are in turn surrounded by "parafollicular" T-cells. (If there is no antigenic stimulus, you'll see only "primary follicles" of sleepy B-cells in the cortex.)
The next time you get to look at a germinal center under the microscope, check out those proliferating B-cells. The sequence from small B-cell to plasma cell is interesting and unsung in most histology courses. You'll need to know this to understand classical acconts of lymphomas:
Within the medullary cords, expect to see a mix of B- and T-cells and plasma cells. The sinusoids are lined by fixed phagocytes.
Despite the elegant pictures in histology books, lymph nodes are seldom "normal", especially in adults.
LYMPHADENITIS: Inflammation of the lymph nodes
ACUTE LYMPHADENITIS described in "Big Robbins" is not much more than the hyperplasia in a reactive node.
Localized lymphadenitis is most often due to a bacterial infection in the area drained by the lymph node.
Really bad cases have polys and even abscess formation within the nodes. The end result will be a scarred-up lymph node. You have one or more of these.
Generalized lymphadenitis suggests a systemic viral infection.
"Mesenteric adenitis", often indistinguishable from acute appendicitis, is caused by Yersinia enterocolitica.
Acute lymphadenitis, since it comes up suddenly and stretches the capsule, is likely to make the node tender.
CHRONIC NON-SPECIFIC LYMPHADENITIS falls in one of three distinctive patterns.
FOLLICULAR HYPERPLASIA (i.e., lots and lots of big follicles) results from longstanding contact with organisms or "other things" that stimulate the B-cells. If perplexed, think of:
{36371} toxoplasmosis; many bugs in a cell
{40654} toxoplasmosis; tissue reaction (lame-looking granulomas)
|
|
|
|
|
PARACORTICAL LYMPHOID HYPERPLASIA (i.e., lots and lots of lymphocytes, including turned-on ones, in the T-cell regions of the cortex -- often easiest to recognize the the presence of prominent blood vessels) results from longstanding contact with organisms or "other things" that stimulate the T-cells. If perplexed, think of
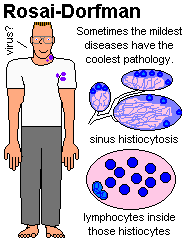
SINUS HYPERPLASIA (formerly "sinus histiocytosis"; i.e., sinusoids with swollen endothelial cells and lots of histiocytes). If perplexed, think of:
|
|
|
![]() Rosai-Dorfman
Rosai-Dorfman
S100 for dendritic macrophages
Wikimedia Commons
MIXTURES OF THE ABOVE cause diagnostic problems. In all the above, capillary endothelial cells are likely to be hyperplastic (rare in cancer).
The most common cause of "unexplained" lymph node enlargement, especially in the groin: DERMATOPATHIC LYMPHADENITIS, melanin and sebum-laden nodes draining chronically inflamed skin. You're likely to see a mix of reaction types.
{35609} dermatopathic lymphadenitis (the red-brown is melanin, the white is sebum)
WARNING: Any of these patterns can be (and occasionally is) mistaken for malignant lymphoma by the inept. Note that the finding of mitotic figures or necrosis doesn't necessarily point to malignancy, while the presence of a variety of cell shapes actually suggests a benign diagnosis. Know your pathologist, and ask for consultation if you are in doubt.
GRANULOMAS
Granulomas with central CASEOUS NECROSIS are probably tuberculosis, some other mycobacterial infection (atypical mycobacteria, leprosy)
Well-made granulomas with NOTHING else are probably sarcoidosis. Also remember Crohn's, berylliosis, and nodes draining Hodgkin's disease.
Granulomas with PUS in their centers are probably caused by one of the
following: (1) lymphogranuloma
venereum![]() ,
(2) cat scratch fever, (3) brucellosis,
(4) plague
,
(2) cat scratch fever, (3) brucellosis,
(4) plague![]() ,
(5) tularemia
,
(5) tularemia![]() ,
(6) glanders-melioidosis, and
(7) other yersinia infections. If you can find none of these, consider (8) X-linked chronic
granulomatous disease (the neutrophil dysfunction problem).
,
(6) glanders-melioidosis, and
(7) other yersinia infections. If you can find none of these, consider (8) X-linked chronic
granulomatous disease (the neutrophil dysfunction problem).
* Granulomas with central necrosis with much karyorrhexis but no pus: Kikuchi-Fujimoto. See below.
|
|
|
|
You have probably already seen the Warthin-Finkeldey giant cells of measles within germinal centers. They show variable immunologic markers -- B-cell, T-cell, and/or dendritic macrophages. Some have intranuclear measles virus inclusions; some do not.
* KIKUCHI-FUJIMOTO NECROTIZING HISTIOCYTIC LYMPHADENITIS (J. Am. Acad. Derm. 59: 130, 2008; Am. J. Clin. Path. 131: 174, 2009): Nobody knows the cause of what seems to be a viral illness (the herpes family exonerated Arch. Path. Lab. Med. 131: 604, 2007); the molecular biology is not that of a lymphoma: Am. J. Clin. Path. 117 627, 2002. Nepalese study: Arch. Path. Lab. Med. 127: 1345, 2003. Big reviews Am. J. Clin. Path. 122: 141, 2004; Arch. Path. Lab. Med. 134: 289, 2010; Medicine 93: 372, 2014. It looks like lupus in the lymph node, but there's lots more cytotoxic CD8+ than CD4+ T-cells. I've got a story about this I'll tell you personally.
|
|
LYMPHADENOPATHY is a clinician's word for a big lymph node.
NON-HODGKIN'S LYMPHOMAS: By definition, monoclonal, malignant tumors of the B- or T-cells, and not of plasma cells, and not Hodgkin's disease. Update Lancet 380: 836 & 848, 2012. By custom, soft tumors of monocytes are included here because they look similar.
These are the common primary tumors arising in the lymphoid tissue (lymph nodes, tonsils, adenoids, spleen, Peyer's patches, non-epitehlial thymus) and there are some special cases. (We cover CNS lymphomas in the "neuro" section; the outlook for primary CNS lymphoma is still "dismal": Cancer 110: 1803, 2007.)
There are about forty kinds at most recent count, each with its own personality. Together, the non-Hodgkin's lymphomas are common. Unlike Hodgkin's, they tend to be widely distributed at the time of diagnosis and to pop up unpredictably as the illness progresses. (The exception is the marginal-zone lymphoma subtype that stays confined to areas of chronic inflammation.) Update, with a focus on molecular markers: Br. Med. J. 362: 139, 2003; also Lancet 362: 139, 2003; J. Clin. Path. 58: 561, 2005.
Most pathologists use the 2008 World Health Organization classification. It is elaborate even by WHO standards and only the highlights can be covered here.
|
|
|
|
![]() Follicular lymphoma, spleen
Follicular lymphoma, spleen
AFIP
Wikimedia Commons
The non-Hodgkin's lymphomas are a subject of perennial fascination for pathologists. Making the diagnosis ("benign or malignant?") is often tough, and classifying the non-Hodgkin's lymphomas (hereinafter "lymphomas") was a major international competitive sport through the 1980's.
Today, the ongoing fascination is in the chromosomal translocations that are the primary way in which white blood cells acquire mutations. In most of the leukemias and lymphomas, the genome is usually NOT destabilized.
Students often find this subject especially difficult to understand. Hence, the focus in this section on "Rules".
RULE: All monoclonal proliferations of lymphocytes are best considered malignant. (Some monoclonal plasma cell proliferations might be benign.)
RULE: Most non-Hodgkin lymphomas are somewhat more common in men, with the most pronounced difference probably being T-lymphoblastic lymphoma (> 2:1).
RULE: Black people and children seldom get nodular lymphomas. (* Nodular lymphomas in children and young adults: Am. J. Surg. Path. 37: 333, 2013.)
RULE: A few specific lymphomas have one or more special risk factors (i.e., helicobacter causes the MALT lymphoma of the stomach; gluten enteropathy "sprue" causes a curious pair of T-cell lymphomas: Gastroent. 132: 1902, 2007; Am. J. Clin. Path. 127: 701, 2007).
"Lymphomas of the immunocompromised" are sometimes real neoplasms, sometimes virally-induced hyperplasias.
* Ataxia-telangiectasia (homozygotes, probably heterozygotes) is a risk factor for most lymphomas and lymphoid leukemias.
Chronic hepatitis C virus infection is now recognized as placing patients at increased risk, and eliminating the virus reduces this risk (Am. J. Med. 120: 1034, 2007).
Sjogren's syndrome (J. Imm. 180: 5130, 2008; Blood 111: 4029, 2008) gives 6x the risk for B-cell lymphomas overall; there are several types (MALT, follicular and large B-cell, famously marginal zone lymphoma).
Hashimoto's thyroiditis also places a person at increased risk for B-cell lymphomas (update J. Clin. Path. 61: 438, 2008). Gluten enteropathy / celiac sprue gives an increased risk for T-cell lymphoma. Does effective treatment reduce the risk? Yes! (Dig. Dis. Sci. 53: 972, 2008) No! (Am. J. Med. 115: 191, 2003).
The increased risk to rheumatoid arthritis patients, also very well-known (Arth. Rheum. 48: 963, 2008 confirmed this but discredited the idea that non-arthritic relatives are at extra risk).
Environmental risk factors for lymphoma are poorly-understood; currently there's an interest in herbicides and pesticides (I think it could be real but a relatively minor risk -- Am. J. Epidem. 147: 891, 1998, Occup. Environ. Med. 60: E11, 2003; Acta Haem. 116: 153, 2006; "only chlordane" Canc. Ep. 15: 251, 2006 from the NIH; Env. Health Perspect. 111: 179, 2003 -- any link to persistent organochlorides must be weak; others) and hair-coloring agents (U.S.; review Cancer Inv. 18: 467, 2000 & Cancer Causes & Control 10: 617, 1999 from the FDA; relationship if any is clearly weak; Am. J. Pub. Health 88: 1767, 1998 no animal model), as well as the African poinsettia (Burkitt's).
RULE: At surgery or autopsy, lymphoma tissue feels like "fish flesh" (i.e., there is very little fibrosis) or "firm rubber" (i.e., there is some fibrosis but not much).
RULE: Fatigue, malaise, night-sweats, fever, and weight loss are the usual symptoms (if any) of these diseases. These are called the "B symptoms" used in staging. The cause, which must involve cytokines, has proved remarkably elusive.
A significant number (in some series, as many as half) of patients with "fever of unknown origin" prove to have non-Hodgkin's or Hodgkin's lymphoma.
RULE: A majority of lymphomas arise in the lymph nodes (one or more groups). Several groups of nodes may pop up at once. Nodular lymphomas almost always arise in lymph nodes.
RULE: A large minority arise in extra-nodal lymphoid tissue, i.e., Waldeyer's ring, stomach, terminal ileum, skin, marrow.
RULE: When lymphomas arise in lymph nodes, they present as non-tender enlargement.
RULE: Lymphomas metastasize to other lymphoid tissues (nodes, spleen, etc.), and eventually to the marrow, blood ("leukosarcoma", less often "lymphemia") and other organs. Low-grade lymphomas metastasize as small nodules, while high-grade lymphomas metastasize as bulky masses.
RULE: Mitotic figure counts tell the growth rate of a lymphoma, but unless the mitotic figures are bizarre, they do not help distinguish it from a benign lymph node. (Have you ever "counted mitoses" in a normal germinal center? Try it!)
RULE: The lower the grade of the lymphoma, the MORE likely the bone marrow is to be involved at the time of diagnosis. Paradoxical, no?
RULE: Lymphomas tend to spread to sites according to their B-cell or T-cell origin. B-cell tumors go to the germinal centers, their mantles, and the outsides of the splenic white pulp. T-cell tumors to the anterior mediastinum, paracortical regions of nodes, insides of splenic white pulp, etc. Skin lymphomas are usually of T-cell origin.
RULE: The malignant cells of lymphomas are MORE uniform than the mix of cells normally seen in lymphoid tissue, and they recapitulate some phase in the life history of either normal B-cells or T-cells. Don't expect to see much "cytologic atypia" in a lymphoma. Remember that the genome is usually not destabilized in lymphomas. (Especially, immunoblastic lymphomas can look pretty wild.)
RULE: Lymphomas that grow as nodules within a lymph node ("trying to be germinal centers") are called NODULAR or FOLLICULAR (synonyms). They are always of B-cell origin, and the lymphoma cells will closely resemble one of the forms in the sequence from resting B-lymphocyte to plasma cell.
{23581} nodular lymphoma
RULE: Nodular lymphomas tend to be indolent lesions with natural histories that are relatively unaffected by classical chemotherapy. Historically, they have been incurable, though this seems to be changing. Each nodular lymphoma has a better prognosis than its diffuse counterpart, and is likely to transform into it sooner or later. This makes sense, since follicle formation is a sign of good differentiation. Less often, a nodular lymphoma transforms into a diffuse large-cell or immunoblastic lymphoma.
* A lymphoma with two different morphologic appearances and genetic clones is a "composite lymphoma". This is fairly common, and may represent transformation of one lymphoma into another by additional mutations, or two separate malignant tumors. Getting it worked out: Am. J. Clin. Path. 99: 445, 1993; Am. J. Path. 154: 1857, 1999; NEJM 341: 764, 1999. The most familiar (CLL/WDLL mixed in with follicular lymphoma) is two different clonal tumors (Am. J. Clin. Path. 137: 647, 2012).
RULE: Most nodular lymphomas of all kinds feature one of two characteristic translocations, either t(11;14) or t(14;18). Each involves the immunoglobulin heavy-chain region on chromosome 14. This is brought into contiguity either with the bcl1 / PRAD / cyclin D1 oncogene on chromosome 11 or the bcl-2 oncogene on chromosome 18.
![]() Diffuse lymphoma
Diffuse lymphoma
WebPath Photo
![]() Diffuse B-cell lymphoma
Diffuse B-cell lymphoma
Gross and microscopic
Wikimedia Commons
bcl-2 produces a protein on the inside of mitochondria that prevents the cell from undergoing apoptosis.
RULE: Small lymphocytic lymphoma ("well-differentiated lymphocytic lymphoma", "the solid phase of chronic lymphocytic leukemia"), in which the cells perfectly resemble normal lymphocytes, is always diffuse, never nodular.
RULE: The histologic type of a lymphoma is much more important than its stage in determining prognosis. (This is the opposite of Hodgkin's disease.)
RULE: Large, polyclonal, benign proliferations of lymphocytes may occur anywhere there is lymphoid tissue, and have earned the dubious name PSEUDOLYMPHOMA. Distinguishing these from real lymphomas is a challenge.
Also remember that certain autoimmune diseases feature heavy polyclonal lymphoid infiltration of salivary glands (Sjogren's), thyroid (Hashimoto's), islets (type I diabetes), or kidneys (autoimmune interstitial nephritis).
* For some reason, Lyme disease![]() produces pseudolymphomas in the ear lobes. No one has a clue why.
produces pseudolymphomas in the ear lobes. No one has a clue why.
RULE: Pathologists trying to distinguish malignant lymphomas from benign lymph node hyperplasias and pseudolymphomas pay special attention to:
(1) EFFACEMENT OF THE NORMAL LYMPH NODE ARCHITECTURE;
(2) CELL UNIFORMITY ("monotony", suggests lymphoma, but even follicular lymphomas are infiltrated by the same benign cells as grow in a germinal center);
* (3) Presence of macrophages laden with nuclear debris (TINGIBLE BODY MACROPHAGES, a sign that the process is EITHER benign OR a high-grade lymphoma, because in low-grade lymphomas you won't see much apoptosis);
(4) Widespread bcl-2 protein staining is a pretty good sign that this is lymphoma.
![]() Tingible body macrophages
Tingible body macrophages
WebPath Photo
(5) VASCULAR PROLIFERATION (new vessels suggest the process is benign), and;
(6) INVASION of surrounding tissue ("capsular transgression", suggests lymphoma).
(7) NECROSIS (apart from apoptosis) is common in some lymphomas, and of course in necrotizing infections, but uncommon in difficult benign lesions.
(8) If "follicles"/"nodules" are present, the ABSENCE OF A MANTLE of small lymphocytes around the light side of the follicle suggests malignancy.
(9) Today's pathologist, asking "Is this lymphoma?", begins as follows:
If it is apparently made of large lymphoid cells, the pathologist will order a CD45 (leukocyte common antigen, positive in lymphomas), a few other lymphocyte markers, cytokeratins (negative in lymphomas), and a few melanoma markers (negative in lymphomas).
![]() Lymphoma in lymph node
Lymphoma in lymph node
Invasion of surrounding fat
Tom Demark's Site
(10) We also want DNA studies for the TYPICAL GENE ARRANGEMENTS, both for diagnosis and to look for residual disease.
{09040} electron micrograph of a malignant lymphoid cell. Note the lack of distinguishing features.
* RULE: Lymphomas in the liver generally center on the portal areas. This also applies to Hodgkin's disease.
RULE: Most lymphomas (Hodgkin's and non-Hodgkin's) may cause generalized dysfunction of benign B-cells (hypogammaglobulinemia), with resulting tendency to infection.
CLASSIFICATION SCHEMES:
Anyone using the terms "lymphosarcoma", "giant follicular lymphoma", or "reticulum cell sarcoma" in today's medicine is terribly out of date.
* THE 1966 RAPPAPORT CLASSIFICATION is archaic but still popular. It was based on certain incorrect (but once-useful) assumptions about the nature of the cells seen in these lesions:
"Well-differentiated lymphocyte"... looks like a normal resting lymphocyte
"Poorly-differentiated lymphocyte"... doesn't look like a normal resting lymphocyte, but is smaller than an endothelial cell
"Histiocyte"... bigger than an endothelial cell, and has lots of cytoplasm
"Undifferentiated cell"... bigger than an endothelial cell, and has only a little cytoplasm
Lymphomas were further sub-divided into "nodular" and "diffuse", depending on their growth pattern. Despite its limitations, the Rappaport system was useful as lymphomas were being sorted out.
* THE 1974 LUKES-COLLINS CLASSIFICATION was based on primitive immunotyping and closer examination of the morphology of the cells, which were compared to those in the centers of normal germinal follicles. Activated-type B-cells from small-cleaved through large-noncleaved cells were appropriately called FOLLICULAR CENTER CELLS. Even more exciting were the IMMUNOBLASTS, very big round cells with very big round nuclei bearing in their centers a single very big nucleolus (I call them "eyeball cells").
* THE 1982 WORKING FORMULATION was a consensus of experts based only on morphology. It worked nicely until it was superseded by the Revised European-American system. You'll still find people using these terms.
LOW GRADE LYMPHOMAS (untreated survival has historically been is around 10 years)
Small lymphocytic
Small lymphocytic, plasmacytoid
Follicular, small cleaved cell
Follicular, mixed small-cleaved and large cell
INTERMEDIATE GRADE LYMPHOMAS (untreated survival has historically been around 5 years)
Follicular, large cell
Diffuse, small cleaved cell
Diffuse, mixed small-cleaved and large cell
Diffuse, large cell
HIGH GRADE (quick death untreated, but we have been curing these with classical chemotherapy since the 1970's)
Large-cell immunoblastic (B- or T-cell)
T-Lymphoblastic
Small noncleaved cell (Burkitt's, etc.)
MISCELLANEOUS
Mycosis fungoides / Sézary syndrome
Adult T-cell leukemia/lymphoma with HTLV-1
MATURE T-CELL AND NATURAL KILLER (NK) NEOPLASMS
T-cell prolymphocytic leukemia
T-cell large granular lymphocytic leukemia (this "CLL" variant causes early anemia / marrow burnout; update Am. J. Clin. Path. 136: 289, 2011;
common mutation in STAT3 drives it NEJM 366: 1905, 2012)
Aggressive NK cell leukemia
Adult T-cell leukemia/lymphoma
Extranodal NK/T cell lymphoma, sinonasal type
Enteropathy-type T-cell lymphoma
Hepatosplenic T-cell lymphoma
Blastic NK cell lymphoma
Mycosis fungoides / Sezary syndrome
Primary cutaneous CD30-positive T-cell lymphoproliferative disease
Angioimmunoglastic T-cell lymphoma
Peripheral T-cell lymphoma, unspecified
Anaplastic T-cell large-cell lymphoma
HODGKIN'S
* The immunomodulators (anti-TNF-alpha agents, anti-CD11a agents, anti-interleukin-2 receptor / CD25 agents, anti-interluekin-1 receptor agents) probably do NOT result in an overall increase in lymphoproliferative disease despite the WHO; however, people on these medicines are prone to develop lymphoid hyperplasias (Mod. Path. 22: 1532, 2009).
Here are the most common ones:
SMALL LYMPHOCYTIC LYMPHOMA (SLL, "well-differentiated lymphocytic lymphoma", WDLL, "the solid phase of chronic lymphocytic leukemia")
This B-cell lymphoma is composed of cells that look like never-stimulated, resting lymphocytes, of the sort seen adjacent to germinal centers. They look normal but don't work. (* Maybe this is why this lymphoma never forms nodules.)
{23575} small lymphocytic lymphoma. There is a small vessel running across the picture. Use the endothelial cell nuclei to gauge the sizes of cells.
The bone marrow is always involved at the time of diagnosis, and if the cells spill into the bloodstream (an absolute lymphocyte count over 5000), "chronic lymphocytic leukemia" is said to be present. See below.
Patients are generally older adults. Despite systemic involvement, the disease progresses very slowly, and seldom kills.
Around 30% of these patients eventually develop a more aggressive B-cell lymphoma (including 1% who get a very aggressive one, i.e., RICHTER'S SYNDROME), as in CLL.
{23854} CLL, transforming into a more aggressive cancer. Note the numerous small lymphocytes and the blasts.
![]() Well-differentiated lymphocytic lymphoma
Well-differentiated lymphocytic lymphoma
Tom Demark's Site
LYMPHOPLASMACYTIC LYMPHOMA (Am. J. Clin. Path. 136: 195, 2011) features cells with slightly more abundant, purple cytoplasm and production of monoclonal paraproteins. As a rule, these diseases are somewhat more aggressive than generic small cell lymphocytic lymphoma, and they usually produce a paraprotein.
WALDENSTROM'S MACROGLOBULINEMIA produces large amounts of IgM pentamers. In addition to the problems seen in any lymphoma, patients suffer with hyperviscosity syndrome (nosebleeds / bleeding gums / bleeding from other mucosal surfaces; dizziness / headache / other neuro problems; eye problems; maybe other problems; look for "sausage link" retinal veins). Like "regular small lymphocytic lymphoma", This is a disease of the elderly.
It tends to be indolent and requires therapy only when the blood becomes too viscous; after treatment, the malignant cells stay around but may not cause further troubles (Am. J. Clin. Path. 135: 365, 2011).
* Future pathologists: Look in the nuclei for "Dutcher bodies", masses of IgM (similar to the familiar "Russell bodies", but in the nucleus). These let you be confident that you're looking at lymphoma. Transformation into a more aggressive cancer can supervene as in the more familiar small lymphocytic lymphoma.
* New suggested criteria for Waldenstrom's: Am. J. Clin. Path. 116: 420, 2001.
* While Waldenstrom's is the most common cause of the hyperviscosity syndrome, you may also see it in some plasma cell myeloma patients, or when there are too many red cells (usually in polycythemia vera), or too many white cells (usually in chronic myelogenous leukemia), or one of the diseases with way too many chylomicrons, or with a cryoglobylin or very bad polyclonal gammopathy, or with way or too many platelets (the reasons are complicated, involving platelet-endothelium interactions, and unlike the others, the blood probably won't be hyperviscous in the lab).
|
|
ALPHA HEAVY-CHAIN DISEASE (now moved along with the other heavy-chain diseases into the plamsa-cell neoplasms section of the W.H.O. classification) typically affects the small bowel and is fairly common in the Near-East. Most victims are young adults, who present with malabsorption.
{13673} heavy chain disease; plasmacytoid cells in intestinal mucosa
* This transforms into the aggressive "Mediterranean abdominal lymphoma", a B-cell immunoblastic lymphoma.
{19504} Mediterranean lymphoma, small bowel
GAMMA HEAVY-CHAIN DISEASE is a marker for a more aggressive lymphoma that generally affects the elderly. Look for big tonsils.
MU HEAVY CHAIN DISEASE generally turns leukemic early.
DIFFUSE LARGE B-CELL LYMPHOMA
The most common of the non-Hodgkin's lymphomas.
Abnormalities of BCL-6 on 3q27 is the most common genetic abnormality. Today these are subdivided by their molecular pathology (if you're asked, it's about BCL6, BCL-2, MYC).
Before genetic profiling, we knew that CHOP chemotherapy has cured around 40-50% of these lymphomas. Trying to sort out which ones responded was one of the first successful applications of microarray gene expression technology (Nature 403: 503, 2000; Nat. Med. 8: 68, 2002). For practical work, there are six genes whose expression tell the prognosis (NEJM 350: 1828, 2008; update PNAS 105: 13520, 2008).
Watch for discovery of the mutations underlying this variability. CARD11 as an oncogene: Science 319: 1656, 2008.
* This lymphoma is so common that even the ophthalmologists have a series of cases starting in the orbit (Am. J. Ophth. 154: 87, 2012).
* These can pop up in the stomach, and if helicobacter is present, eradicating it can cure the tumor, just as in the more familiar MALTomas -- Blood 119: 4838, 2012.
MANTLE CELL LYMPHOMA (Hum. Path. 31: 7, 2002; Arch. Path. Lab. Med. 132: 1346, 2008)
It always features t(11;14), involving cyclin D1 (bcl-1, CCND1) (assay Am. J. Path. 154: 1449, 1999; Blood 93: 1372, 1999; cyclin D1 is easy to stain for in sections), which is brought adjacent to an enhancer of the IgH gene.
It's a disease mostly of older men, and often arises extranodally. It is quite aggressive and hard-to-treat. Good results are now being reported (Am. J. Clin. Path. 137: 634, 2012), especially with bortezomib substituted for vincristine in R-CHOP (NEJM 372: 944, 2015).
* Don't worry about the details for pathologists. There are extra-aggressive "blastoid" and "pleomorphic" subtypes, etc., etc.
MALT LYMPHOMA ("maltoma", "extranodal marginal zone B cell lymphoma"; "marginal zone lymphoma of mucosa-associated lymphoid tissue"; named for its occurrence on on mucosal surfaces, of course) is now defined by its a trademark translocation t(11;18) and fusion protein IAP2(API2)/MALT1; AM. J. Path. 162: 1113, 2003, or translocation t(14;18 -- MALT1/IgH) or one of the related translocations involving MALT1.
Many (but by no means all) Hashimoto and Sjogren-associated lymphomas are MALT type.
* Since the MALT lymphoma cells have the markers of B-cells that are just learning to fight a specific antigen, it makes sense that the presence of the antigen keeps the lymphoma going. Someone else can explain the molecular biology. * It's not uncommon for a person to have several MALT-omas in different organs; they are often genetically different, even with different translocations (Am. J. Clin. Path. 134: 112, 2010).
MARGINAL ZONE LYMPHOMA ("marginal cell lymphoma"; Am. J. Clin. Path. 117: 698, 2002)
Like mantle lymphomas, it tends to grow around benign germinal centers.
Dermatopathologists are especially familiar with these, as they tend to arise in the skin at sites of ongoing immune activation -- infamously Lyme disease acting as a promoter (Histopathology 37: 501, 2000).
FOLLICULAR LYMPHOMAS (CB/CC lymphoma)
Formerly divided into "small-cleaved", "mixed small-cleaved and large cell" and "large-cell" subcategories, it's now pretty clear that most of these lymphomas are mixtures of centroblasts (large B-cells with non-cleaved nuclei) and centrocytes (cleaved B-cells) -- both resemble cells in the germinal centers. They grow as nodules surrounded by benign lymphocytes (mostly T-cells).
The "small cells" / centrocytes look like normal lymphocytes except for one or more clefts up the nucleus ("buttock cell", etc.), and they lack the marbly heterochromatin.
The World Health Organization has a grading system:
The pathologist will also report what percentage of the tumor has the follicular pattern. "Follicular" means more than 75%; "follicular and diffuse" means 25-75%; "focally follicular" means less than 25%; "diffuse" means no follicles.
Patients are usually in middle-aged or older. The bone marrow is usually involved at the time of diagnosis. The translocation t(14;18), with bcl2 brought next to IgH, is usual.
If only a mass is discovered and there are no symptoms, most physicians today recommend doing nothing; it takes an average of 3 years for symptoms to appear. Today they are easy to treat initially, but tend recur repeatedly after treatment begins, each time sooner than last.
About half of these transform into a diffuse B-cell lymphoma; the transformation is often accompanied by generalized symptoms (perhaps for the first time) and the disease becomes harder to manage.
{23599} mixed lymphoma; use the endothelial cell at 2:30 as a size marker
{23683} mixed lymphoma
{23596} nodular large-cell; at this power, just appreciate the nodularity
{23581} nodular lymphoma
Telling nodular lymphomas from hyperplastic germinal centers:
MORE LYMPHOMAS
{23590} diffuse small cleaved lymphoma (all
you can tell is that it is small cleaved)
{23593} diffuse small cleaved lymphoma (all you can
tell is that it is diffuse)
{46344} diffuse small cleaved lymphoma, marrow
{23581} nodular lymphoma
|
|
|
ANAPLASTIC (T-CELL) LARGE CELL LYMPHOMA (Arch. Path. Lab. Med. 135: 19, 2011) is rather less aggressive than the other large ones. The famous "hallmark cells", with multilobular, horseshow-shaped nuclei are a distinctive feature.
The more familiar form of this cancer also features t(2;5) with production of a fusion product oncogene (NPM/ALK, Blood 93: 3088 & 3913, 1999) and is now called "ALK+ / anaplastic lymphoma kinase", a fusion product involving nucleophosin (update Am. J. Clin. Path. 130: 628, 2008.
* Future pathologists: All anaplastic large-cell lymphomas light up with CD30/Ki1.
When primary in the breast, the woman commonly has implants, and perhaps this is the first genuine link between a disease -- though vanishingly rare -- and the implants (JAMA 300: 2030, 2008; Am. J. Surg. Path. 36: 1000, 2012).
{08787} large-cell lymphoma
{15389} large-cell lymphoma
{23647} * "angioimmunoblastic lymphadenopathy",
a T-cell lymphoma with vascular proliferation that we're not going to worry about -- note the vessels and the monomorphic cell infiltrate)
* Some of the larger-celled lymphomas may be indistinguishable on H&E from the rare TRUE HISTOCYTIC LYMPHOMA (an oxymoron; histiocytes are not lymphocytes). The immunostaining and chromosomal studies will clarify everything.
{23674} true histiocytic lymphoma, trust me
EFFUSION LYMPHOMAS, in the body cavities without a solid phase,
are usually seen in immunosuppressed people and always
are caused by KSHV (herpes 8![]() )
(Lancet 346: 883, 1995; Lancet 347: 980 & 1042, 1996).
KSHV is now required for case
definition (Cancer 111: 224, 2007).
)
(Lancet 346: 883, 1995; Lancet 347: 980 & 1042, 1996).
KSHV is now required for case
definition (Cancer 111: 224, 2007).
{10935} lymphoma arising in thyroid; my case
{10937} lymphoma arising in thyroid; my case. Notice that the lymphocytes are growing within a
follicle.
|
|
|
|
|
{00245} immunoblastic lymphoma
{10691} immunoblastic lymphoma, cytology
{10724} immunoblastic lymphoma
{10772} immunoblastic lymphoma
{23623} immunoblastic lymphoma
{23689} immunoblastic lymphoma
{08017} lymphoma in the heart
{11630} lymphoma in the pericardial space
{11633} lymphoma, primary in the heart
{20227} lymphoma, primary in the stomach
{15446} lymphoma, primary in the stomach
{15542} lymphoma, primary in the stomach
T-LYMPHOBLASTIC LYMPHOMA
This is the most important pediatric lymphoma (typically a teenaged's guy's disease); it is the solid counterpart to T-cell acute lymphoblastic leukemia.
* These smallish T-cells have convoluted (i.e., more than one cleft) nuclei, though they are not as complex as in Sézary syndrome (below). Immunologists note similarities with baby, intra-thymic T-cells. The usual t(14;21) and its molecular biology: Proc. Nat. Acad. Sci. 97: 3497, 2000. There is often a gain-of-function mutation of NOTCH: Nat. Med. 13: 1203, 2007.
In keeping with its thymocyte origin, it typically presents itself in the anterior mediastinum (i.e., thymus area).
The prognosis has historically been not-so-good. Try a new chemotherapy protocol.
{00242} T-lymphoblastic lymphoma. Trust me.
BURKITT'S LYMPHOMA ("small non-cleaved cell lymphoma", * one of Rappaport's "undifferentiated lymphomas"; Lancet 379: 1234, 2012)
A famous B-cell tumor, the most common childhood cancer where malaria is endemic. It is the fastest-growing human solid tumor. Most often, the childhood variant arises in the jaw.
{46189} African Burkitt's
{49035} African Burkitt's
![]() Burkitt's
Burkitt's
Section
Wikimedia Commons
![]() Burkitt's
Burkitt's
Smear
Wikimedia Commons
The Epstein-Barr virus![]() is part of the cause, but
obviously not the whole story. These tumors also have
a famous translocation that places the oncogene myc on chromosome 8 under the control of the
IgH regulator on chromosome 14. (* Less often, myc joins the kappa chain gene on
2, or lambda on 22).
is part of the cause, but
obviously not the whole story. These tumors also have
a famous translocation that places the oncogene myc on chromosome 8 under the control of the
IgH regulator on chromosome 14. (* Less often, myc joins the kappa chain gene on
2, or lambda on 22).
NOTE: We've already seen that many lymphomas in immunosuppressed patients, both inside and
outside the CNS, are strongly linked to the Epstein-Barr virus![]() .
Many, but not all, have Burkitt-like histopathology.
Nowadays we call these
"post-transplantation lymphoproliferative disorders", and they tend to
regress if immunosuppression can be discontinued.
.
Many, but not all, have Burkitt-like histopathology.
Nowadays we call these
"post-transplantation lymphoproliferative disorders", and they tend to
regress if immunosuppression can be discontinued.
![]() Epstein-Barr
Epstein-Barr
Post-transplantation lymphoproliferative disorder
WebPath Photo
The lymphoma cells are strikingly uniform, with big blue nuclei, and deep blue cytoplasm laden with lipid droplets. Tingible body macrophages loaded with this lipid appear as white "stars" against the blue "sky".
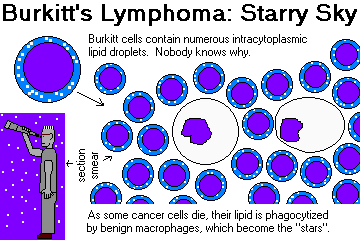
The "starry sky" appearance of Burkitt's is a favorite exam question. Just to confuse you, tingible body macrophages appear as similar "stars" against the not-so-blue-as-Burkitt's "sky" of a normal lymph node. Despite "Big Robbins", the stars of Burkitt's are more conspicuous than other tingible-body macrophages because they are heavily laden with lipid.
AFRICAN BURKITT'S, almost always EBV-positive, is generally curable with chemotherapy, if you can get it to the victims.
By contrast, AMERICAN BURKITT'S, a sporadic disease of young people and the immunocompromised, may be EBV-positive or EBV-negative. It can produce masses most anywhere, and has a worse prognosis.
MYCOSIS FUNGOIDES / SÉZARY SYNDROME (Lancet 371: 945, 2008.
Lymphomas of the epidermis and upper dermis, composed of large T4-cells with very elaborately infolded ("cerebriform") nuclear membranes. The distinctive "Pautrier microabscesses" (misnamed) are clusters of these T-cells within the epidermis (* they're great for exams but in real life, you'll only see them in 25% or so of MF cases; the illness is now diagnosed by criteria.)
In "mycosis fungoides" (Latin for "Toadstools! Toadstools!"), patients suffer from red, peeling skin for some years, then enter a plaque and eventually a tumor phase, in which the patient looks horrible and has lymphoma throughout the body. The transformation is to a large cell lymphoma.
{40003} mycosis fungoides
{40004} mycosis fungoides
{12747} mycosis fungoides, plaque phase
{12751} mycosis fungoides
{12754} mycosis fungoides
{13117} mycosis fungoides
{13781} mycosis fungoides
{13784} mycosis fungoides
{24740} mycosis fungoides, histopathology; note Pautrier microabscesses
{12759} mycosis fungoides, Pautrier microabscesses
{13793} mycosis fungoides, Pautrier microabscess
{13796} mycosis fungoides cells in a lymph node (look
how wiggly the nuclear membranes are)
{09042} mycosis fungoides cell, electron micrograph
In "Sézary syndrome", the red skin does not transform into tumors. Instead, the cells circulate in the blood as a leukemia. To make the call, there must be >1000 Sezary cells in the blood, and they must be an abnormal clone of T-cells and there must be lymph node involvement. It overlaps with mycosis fungoides. The disease is slowly progressive, and survival for several years is usual.
{12757} Sézary patient
{16544} Sézary cell
{23722} Sézary cell
{15409} Sézary cell
ADULT T-CELL LEUKEMIA-LYMPHOMA
A rare, very aggressive malignancy of T-helper cells.
It is strongly linked to the HTLV-I retrovirus, which is transmitted like AIDS, binds to the same receptor (CD4), is neurotrophic, and lies dormant for a long time. The leukemic cells carry the provirus.
We now check all donor blood for this virus.
* The malignancy is preceded by polyclonal T-cell hyperplasia, due to induction of T-cell IL-2 receptors by the virus.
* Have a pathologist show you the distinctive "flower cell" lymphocytes in the blood in the leukemia.
* Hypercalcemia is common in this disease; the molecular biology is curious, and involves the leukemic cells turning into osteoclasts (Blood 99: 634, 2002).
The disease (like the virus) is more common in Japan and the Caribbean. HTLV-I in Japan: Lancet 343: 213, 1994.
* Don't worry about the cancers of monocyte-macrophage origin. MALIGNANT HISTIOCYTOSIS ("histiocytic medullary reticulosis"), a very aggressive, fortunately rare cancer of blood-cell-eating true macrophages, is worth mentioning here. Not a cancer, but also deadly.... HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS, a sometimes-genetic (often perforin), sometimes-acquired (triggered by infection or rheumatologic disease: Pediatrics 118: e216, 2006; Blood 106: 3090, 2005; South. Med. J. 100: 208, 2007) illness. The macrophages go crazy, along with the CD8 cells, and destroy the body (Am. J. Clin. Path. 137: 786, 2012; Mayo Clin. Proc. 89: 484, 2014).
{23668} malignant histiocytosis with erythrophagocytosis
|
|
HODGKIN'S DISEASE ("Hodgkin's lymphoma"; J. Clin. Path. 55: 162, 2002)
A common (7500 cases/year in the U.S.), usually-curable cancer that typically affects young adults. (There is a second peak in older adults; their disease tends to be more aggressive.)
Hodgkin's always presents as a mass in one of the areas where there's already a lot of lymphoid tissue, most often the neck or mediastinum. Risk factors are ill-defined, and "epidemics" could perhaps be statistical accidents. Family members are at several times increased risk, and a monozygous twin is at 100 times the base risk (NEJM 332: 413, 1995).
A previous history of Epstein-Barr![]() infectious mononucleosis
supposedly triples one's risk for Hodgkin's
disease. This has held up and having Epstein-Barr virus on board
places one at an increased risk, but exactly what the relationship is remains
obscure (possible mechanisms: Blood 106: 4345, 2005). Of course,
many Hodgkin's patients are EBV-negative (Blood 106: 2444, 2005).
infectious mononucleosis
supposedly triples one's risk for Hodgkin's
disease. This has held up and having Epstein-Barr virus on board
places one at an increased risk, but exactly what the relationship is remains
obscure (possible mechanisms: Blood 106: 4345, 2005). Of course,
many Hodgkin's patients are EBV-negative (Blood 106: 2444, 2005).
* Hodgkin's disease is rare in the Orient. For some reason, pediatric Hodgkin's is common in the poor nations.
* Hodgkin's disease is a recognized complication of AIDS, though less typical than non-Hodgkin's lymphomas. Not surprisingly, AIDS patients with Hodgkin's disease tend to lack lymphocytes (Cancer 67: 1865, 1991).
The malignant cell is the REED-STERNBERG CELL, but until the late stages of the disease, the tumor masses are composed primarily of inflammatory cells responding to the cancer.
You must recognize the CLASSIC REED-STERNBERG CELL:
|

|
{23560} Reed-Sternberg cell
{20057} Reed-Sternberg cell
{36398} Reed-Sternberg cell, not H&E; cytology
{36401} Reed-Sternberg cell, not H&E; cytology
{40423} Reed-Sternberg cell, mitosis
|
|
|
|
Everybody accepts the 1965 RYE CLASSIFICATION of Hodgkin's disease with the W.H.O. modification that separated-out nodular lymphocyte predominant.
NODULAR LYMPHOCYTE PREDOMINANT:
In this illness, there are sheets of lymphocytes and some RS-like cells. However, the RS cells don't even immunostain like in other forms of Hodgkin's (they are CD15-, CD45+, B-cell markers are positive), and it's probably "not really Hodgkin's, maybe a dysplasia": old work Blood 87: 2428, 1996; Am. J. Path. 146: 812, 1995; different mutations Blood 101: 706, 2003; long-term follow-up supports the good prognosis but a few patients transform to a diffuse large B-cell lymphoma (Cancer 116: 631, 2010).
The main reason to "type" Hodgkin's is to rule this in or out, since it's noted for late recurrence (NEJM 318: 214, 1998).
CLASSICAL LYMPHOCYTE PREDOMINANT / "LYMPHOCYTE RICH": A background of normal, monotonous, small lymphocytes.
{46338} Lymphocyte predominant Hodgkin's
{46339} Lymphocyte predominant Hodgkin's
Reed-Sternberg cells of any kind may be rare! See NEJM 319: 246, 1988.
This variant generally announces itself in a single group of nodes, and almost all patients get cured by today's therapies.
Don't diagnose "chronic lymphocytic leukemia" or "small lymphocytic lymphoma" in a young person until you've sectioned through the block in your search for the diagnostic cell.
MIXED CELLULARITY: There are many Reed-Sternberg cells and variants, in a background of lymphocytes, plasma cells, eosinophils, and histiocytes. This variant can present at any stage.
{23539} mixed cellularity Hodgkin's disease
{46342} mixed cellularity Hodgkin's disease
{46343} mixed cellularity Hodgkin's disease
LYMPHOCYTE DEPLETION: Mostly cancer cells, little else. Probably the Hodgkin's cells have taken additional mutations.
* The background may be lots of poorly-woven collagen ("diffuse fibrosis variant") or just reticulin ("reticular variant"), with wildly anaplastic cells.
The disease often (but not always) presents at late stage.
Future pathologists: You won't make this diagnosis unless there's a recognizable Reed-Sternberg cell or a previous diagnosis of Hodgkin's disease.
{23524} lymphocyte depleted Hodgkin's disease. Just plain anaplastic.
NODULAR SCLEROSIS: This features lacunar Reed-Sternberg variants and a tendency for the lesion to become crisscrossed by dense collagen bands. The prognosis is generally good.
{23542} nodular sclerosing Hodgkin's disease
{23545} nodular sclerosing Hodgkin's disease
{23548} lacunar Reed-Sternberg variants
{23551} lacunar Reed-Sternberg variant
NOTE: There are subtypes of each common type....
Sex ratios: Nodular sclerosis is a bit more common in women. All the other forms are more common in men.
Having described this elegant classification scheme, I am almost sorry to have to add that the prognosis for any particular case of Hodgkin's disease is determined by stage, rather than by type. Almost all patients with stage I or IIA disease are now cured. This drops to around 50% for patients presenting at stage IV.
Lymphocyte predominant presents at low stage, mixed cellularity at low or high stage, lymphocyte depletion presents at high stage, and nodular sclerosis is often a mediastinal mass. These differences account for "different prognosis for different Hodgkin's types".
* Update on treating Hodgkin's, including recognizing people with good prognosis for whom the most intensive therapies can be avoided (Blood 120: 822, 2012).
REED-STERNBERG VARIANTS are also malignant.
MONONUCLEAR REED-STERNBERG-LIKE CELLS ("Hodgkin cells") have single-lobed nuclei and one nucleolus. They may be seen in any variant of Hodgkin's disease.
* LP CELLS (* "L&H cells") have scanty cytoplasm, big knobby nuclei, and small nucleoli. They are seen in lymphocyte predominance Hodgkin's disease.
LACUNAR REED-STERNBERG CELLS have abundant, pale cytoplasm (* an artifact of formalin fixation). They are seen in nodular sclerosis Hodgkin's disease.
POLYLOBULATED REED-STERNBERG CELLS ("popcorn cells") look like good Reed-Sternberg cells, except that the nucleoli aren't so impressive. They are typical of mixed cellularity Hodgkin's disease.
* PLEOMORPHIC REED-STERNBERG CELLS are anaplastic versions of the familiar form. They make up the bulk of the tumor in lymphocyte depletion Hodgkin's disease.
REED-STERNBERG CELL RULES:
While a classic Reed-Sternberg-like cell may appear in other diseases (even "infectious mono"), its presence in the proper background (see below) gives the diagnosis of Hodgkin's disease.
You must see a CLASSIC Reed-Sternberg cell before making the diagnosis.
Hodgkin's begins as an enlarged node or group of nodes. While we do not test you on staging, everybody knows these basics (used for a majority of non-Hodgkin's lymphomas too):
Stage I... one node group or organ
Stage II... one side of the diaphragm
Stage III... both sides of the diaphragm
Stage IV...marrow, or two extra-lymphatic organs
"A" means no systemic symptoms
"B" means fever, weight loss (>10%), or night-sweats.
The classic Hodgkin's fever is the "Pel-Ebstein", or intermittent spiking fever.
{20056} Hodgkin's disease in a cervical node (we
would of course diagnose this only with microscopy)
{46348} Hodgkin's disease in the spleen
{46349} Hodgkin's disease in the spleen
Hodgkin's disease spreads predictably along contiguous groups of lymph nodes. As it spreads, there may be transformation:
Lymphocyte predominant turns into mixed cellularity or lymphocyte depletion.
Mixed cellularity turns into lymphocyte depletion.
Nodular sclerosis generally keeps its type.
When I was in college, I had a classmate who I was told "might end up being the first human cured of Hodgkin's disease." He died, but the fact that Hodgkin's is now cured so often is one of the 20th century's great triumphs. Even chemoresistant Hodgkin's now seems to be getting cured sometimes using a new protocol (with autologous stem cells, Cancer 113: 1344, 2008).
Minor mysteries of medicine:
(1) Hodgkin's patients often notice pain at sites of disease after they drink alcohol.
(2) Hodgkin's patients often have cutaneous anergy, even early in their disease.
INTRODUCING THE LEUKEMIAS
![]() Leukemia / myelodysplasia
Leukemia / myelodysplasia
"Pathology Outlines"
Nat Pernick MD
Leukemia ("white blood"), discovered and named of course by Virchow, is a generic term for replacement of the bone marrow by cancerous blood cells. These usually (but not always; many acute leukemias are initially "aleukemic") are spilling over into the bloodstream; in any case, expect a "packed marrow" except in early CLL.
The story of progress in the treatment of the leukemias, all considered incurable when I was in college, is described in Cancer 113(7S): 1933, 2008.
{23848} packed marrow; * this was late-stage CLL
{36032} packed marrow; * this was AML
{12347} packed marrow; * this was AML
ACUTE LEUKEMIAS ("poorly differentiated leukemias") are overgrowths of cells that fail to mature (BLAST CELLS). These diseases are very aggressive, and cause death in weeks or months.
{16243} blast with Auer rods
{29475} lymphoid blasts, pap stain. Big pale nuclei.
HOW TO SPOT A BLAST: (review from "Histology")
Future pathologists: Despite "Big Robbins", you cannot tell whether a generic, undifferentiated "blast cell" is lymphoid or myeloid without doing special stains and/or chromosomal studies noted above. You'll learn below that Auer rods are sure markers for myeloid differentiation. Of course, today you must use immunohistochemistry (easy approach Arch. Path. Lab. Med. 132: 462, 2008).
These cells are not especially fast-growing, but they fail to mature. Even if they do not "crowd out" their healthy neighbors, they tend to inhibit normal production of other blood cells.
Acute leukemia presents abruptly as one of the cytopenias (anemia, neutropenia, and/or thrombocytopenia). Bone pain is likely to result from expansion of the marrow and infiltration of the periosteum.
Later, involvement of other organs is common. Brain involvement is especially troublesome. T-cell leukemias often produce a mass in the anterior mediastinum (why?).
{23842} acute lymphocytic leukemia, brain
{08734} acute leukemia, liver; as you would expect, the leukemia is blue
{08735} acute leukemia, liver
{08736} acute leukemia, liver
Death typically results from hemorrhage (cerebral, GI, other), and/or infection (neutropenia, chemotherapy), and/or complications of bone marrow / stem cell transplantation.
{06269} fatal cerebral hemorrhage in leukemia
{01735} brain and dura, acute leukemia
The biology of the acute leukemias is very well-studied (still interesting Lancet 349: 118, 1997). The refractory ones have pumps to remove chemotherapy drugs, etc., etc.
By contrast, CHRONIC LEUKEMIAS ("well-differentiated leukemias") feature cancer cells that do mature (more or less), and that have a natural history measured in years.
Like acute leukemia patients, these patients may present with a cytopenia problem. Or they may have problems from a high white ("leukostasis" plugging small vessels), or may notice lymphadenopathy or organomegaly, or the high white count may be an incidental finding on "routine lab".
RULE: Marrow cells (most leukemias, extramedullary hematopoiesis) in the spleen involve the red pulp. Lymphomas in the spleen (at least the B-cell type) involve the white pulp.
RULE: Any leukemia can involve the lymph nodes and make them large, but the enlargement is seldom so spectacular as in Hodgkin's or non-Hodgkin's lymphoma.
RULE: Any leukemia (or lymphoma) can involve the liver, but enlargement is usually not spectacular. Hodgkin's and non-Hodgkin's lymphomas will first appear in the portal areas.
LYMPHOID LEUKEMIAS recall the various kinds of normal lymphocytes, and MYELOID ("myelogenous", or better, "granulocytic") leukemias recall a normal granulocyte.
You remember that MYELOCYTES, the precursors of granulocytes, are the most common cell in normal bone marrow (MYELO-); hence their name.
Cell turnover in the leukemias (and the closely-related polycythemia vera and primary myelofibrosis) is much increased, placing these patients at risk for gout. The risk increases further when cancer cells are dying by the pound during therapy. The thoughtful oncologist gives prophylactic medication.
ACUTE LYMPHOBLASTIC LEUKEMIA ("ALL"; diagnosis Am. J. Clin. Path. 111: 467, 1999)
|
|
This is the familiar "childhood leukemia", with peak age in four year old kids. Adult ALL is less common.
{12410} ALL (all you can tell from the smear is "blasts")
ALL seems to strike at random. Radiation exposure is a known risk factor, Down's syndrome kids are at 15x the normal risk, a patient's identical twin has a 20% risk.
* A curious claim that could be true is the idea that leukemia is an unusual response to one or more as-yet-unidentified viruses met at the wrong age; one finding that seems to be robust is a strong correlation between leukemia rates and the diversity of origins, i.e., new immigrants, in an area (Br. Med. J. 313: 1297, 1996; Lancet 349: 344, 1997.)
Rachel Carson's unfortunate (i.e., false and inflammatory) discussion of leukemia in "Silent Spring" is now history. She was right about the danger of DDT to birds' eggs, and her manipulation of the data on leukemia is a minor blot on the memory of a great science writer. Nobody's perfect. Today, there's no serious discussion of the old alleged link between insecticide exposure and leukemia (* the one paper I could find from the decade 1997-2007, Canc. Ep. 16: 1172, 2007, looks like recall bias).
* Pathologists and oncologists used to subclassify acute lymphoblastic leukemia by blast morphology ("FAB classification"; stands for "French-American-British"):
L1: 85%... Cells <= 2x the diameter of a normal lymphocyte; smooth nuclei; more common in kids
L2: 14%... Bigger cells, lots of clefts, often nucleoli; more common in adults
L3: 1%... Even bigger cells, Liquid Burkitt's lymphoma; t(8;14) and everything
{23746} L1
{23833} L1, special stain (cytoplasm is brown)
{23860} L2
{13982} L2, bone marrow
{23758} L3; note the lipid
{13985} L3
L3 is a distinct entity, but L1 and L2 aren't especially useful. Here's a more-modern immunophenotypic classification:
B-cell... 80%... * CD19+...* several subclasses exist
T-cell... 15%... * "intra-thymic markers, like T-lymphoblastic lymphoma"; chances of a cure are much less than with B-cell disease
Nothing... uncommon today...* markers are negative; there is only HLA-DR.
Apart from the fact that all L3's (Burkitt's) are B-cell tumors, there is little correlation between the two systems.
In 2002, the World Health Organization proposed the following system, which seems to be generally accepted now:
3. Biphenotypic acute leukemia
More for cytogeneticists and prognosticators:
Hyperdiploidy (especially "high hyperdiploidy", >50 chromosomes, all good standard chromosomes though), present in a large minority of B-cell ALL's, confers a good prognosis.
Adults with ALL are often "Philadelphia-positive", and kids can be, too (* the latter confers a bad prognosis: NEJM 342: 998, 2000). Several other bad-prognosis translocations, including the Burkitt t(8:14), and the AML variant of the Philadelphia chromosome, are known.
t(12;21) and del(9p) give a relatively favorable prognosis.
Almost all children with ALL get a complete remission on current therapy. Around 90% get apparent cures on today's regimens (NEJM 366: 1445, 2012).
The prognosis for adults is still guarded. Cure rates for teens are now about as good as for young children (J. Clin. Onc. 29: 386, 2011). Occasionally the disease appears in older adults with less chance of cure.
* Precursor T-cell ALL and MLL gene-rearranged leukemias are exceptions that are more refractory to cure.
{23857} ALL in the liver
{32027} ALL in the liver
{34513} ALL in the brain
{49316} ALL in the kidney
{49343} ALL in the testis
* In 1987, Coby Howard,who had ALL and a relapse, and belonged to a family that did not work and received Medicaid as part of welfare, didn't get his $100,000 bone marrow transplant that had maybe 25% chance of working (J. Clin. Onc. 5: 1348, 1987 is from that era) because of Oregon's health-care rationing, which was intended to spend the Medicaid money where it was most likely to do the most overall public good. Pressure groups (liberal, conservative), of course, had a field-day with Coby's death. During the activity at the legislature that followed, it was pointed out that many working people who did not have health insurance were dying because they lacked access both to transplants and to far more basic services. Taxpayers understandably resent being forced to pay for health benefits for others that they cannot get for their own children. Remember poor Coby's name, and the principle.
ACUTE MYELOID LEUKEMIA ("AML", "acute myelogenous leukemia", "poorly differentiated granulocytic leukemia", "acute non-lymphocytic leukemia", "ANLL", etc.; update Lancet 381: 484, 2013 sorts out the molecules)
This is the common acute leukemia of adults (the incidence increases with age, but young adults are often affected, and occasionally children are affected). There are now at least a dozen molecular types, each with its own personality.
Although most cases are idiopathic, many things are known to increase risk. These include:
The cancer arises out of a background of mutated marrow cells that typically include granulocytic and other (erythroid and/or monocyte precursors). All are likely to show some abnormalities (* good tipoff: megakaryocytes with non-lobulated nuclei).
The FAB classification (Ann. Int. Med. 103: 614 & 620, 1985) is worth knowing at the recognition level (though it is of little interest to practicing oncologists, who are today much more concerned about the molecular lesions):

M0: "minimally differentiated AML"; undifferentiated myeloblasts without myeloperoxidase (* new criteria Am. J. Clin. Path. 115: 876, 2001)
M1: "acute myeloblastic leukemia without maturation"; undifferentiated myeloblasts with myeloperoxidase; just maybe a few Auer rods
M2: "acute myeloblastic leukemia with granulocytic maturation"; the most common; some promyelocytic differentiation, maybe a few Auer rods; * t(8;21) is distinctive, with fusion AML1/ETO
M3: "acute promyelocytic leukemia"; very granular promyelocytes, often many Auer rods, DIC (* from annexin II on the surfaces that activates plasmin: NEJM 340: 944, 1999; not the full story Thromb. Res. 113: 109, 2005); * t(15;17) and * t(11;17) are distinctive, and (KNOW:) involve the vitamin A receptor (RARα; discovery Blood 77: 1418 & 1657, 1991).
All-trans retinoic acid (* "tretinoin") matures these cells: NEJM 324: 1385, 1991; NEJM 27: 385, 1992; Blood 85: 2643, 1995; Blood 94: 39, 1999 (* discovered by the Mainland Chinese, who attributed their success to lack of regulation of human research.... Do any of our "ethicists" want to "refuse to use this data which was acquired immorally"?)
* Retinoic acid plus G-CSF for the difficult t(11;17) subtype: Blood 94: 39, 1999. Diagnostic stain Blood 86: 862, 1995.
Arsenic trioxide (!) was introduced into the treatment of M3 (first positive report NEJM 340: 1043, 1999. Taken in combination with the retinoic acid derivatives, the new protocols produce cures (Nat. Med. 14: 1333, 2008; NEJM 360: 928, 2009; 80% cures NEJM 369: 111, 2013)
M4: myeloid and monocytic differentiation; many have inv(16)/t(16;16); (* a subtype M4eo has eosinophils all over the marrow)
![]() Acute Myelomonocytic Leukemia
Acute Myelomonocytic Leukemia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
M5: monocytic differentiation only; * t(9;11)(p22;q23) is typical of M5a; 9p22 is beta-interferon receptor, while 11q23 is the MLL (myeloid-lymphoid leukemia gene; common in infant and adult leukemias, though not childhood leukemias (Proc. Nat. Acad. Sci. 88: 10735, 1991; Blood 94: 283, 1999) as well as adult AML
* MLL also forms a fusion product with MEN-1 (which is adjacent) and a src-like gene on chromosome 19 (Proc. Nat. Acad. Sci. 94: 2563, 1999) in t(11;19) leukemias; this fusion product inhibits p53's product (Blood 93: 3216, 1999).
* M5a is "acute monoblastic leukemia" with very primitive-looking blasts bearing monocyte markers; M5b is "acute monocytic leukemia" with a dented nucleus and granules.
* "Juvenile myelomonocytic leukemia" is a curious illness that often indicates underlying NF1.
M6: "acute erythroid leukemia"; features of red cell precursors predominate; "Di Guglielmo's erythroleukemia" (* future pathologists: look for d-PAS-positive chunks in the cytoplasm)
M7: "acute megakaryoblastic leukemia"; platelet markers; acute marrow fibrosis (* PDGF effect; reticulin); acute megakaryoblastic leukemia is a childhood leukemia (Am. J. Clin. Path. 122-S: S33, 2004).
Rarities not in the schema include acute basophilic leukemia, acute eosinophilia, and several others.
Researchers expressing ongoing dissatisfaction with the WHO 2002 system (for example, the fact that 40-50% of adults with AML have no cytogeneic abnormality at diagnosis) are looking forward to a future classification based on individual mutations, instead.
Be all this as it may, we begin making the distinction among the many variants of AML from one another, and from ALL, by morphology and by the stains listed earlier in the unit. AUER RODS are red-staining, rod-shaped crystalloids made of primary granules (you can see them especially well if you stain for peroxidase). If present, they prove that a blast is "myeloid" and not "lymphoid".
{12338} AML. Note promyelocyte in center.
{16245} blast, and not much else, M1
{13988} blasts, and not much else, M1
{23830} blast, * chloroacetate-esterase (+), M1
{13940} blast, * chloroacetate-esterase (+), M1
{23839} blast, M1; note nucleolus.
{23770} blasts with a few granules, M2
{13991} blasts with a few granules, M2
{16249} blast with Auer rod, M2
{12344} blast with Auer rod, M2 or maybe M3
{14010} blast with great Auer rods
{23776} blasts with lots of granules, M3
{13994} blasts with lots of granules, M3
{16254} blasts with lots of granules, M3
{10109} blasts with lots of granules, M3
{13997} semi-monocyte like blasts, M4; note indented nucleus and gray cytoplasm
{10133} semi-monocyte like blasts, M4
{13937} non-specific esterase in M4
{23800} monocyte-like blasts in M5
{40449} monocyte-like blasts in M5
{40451} monocyte-like blasts in M5
{23803} non-specific esterase in M5
{14001} monocyte-like blasts in M5
{13928} non-specific esterase in M5
{23949} erythroleukemia, M6. If you don't recognize the malignant cells as red-cell precursors, please
check out a histology book.
{14007} erythroleukemia, M6
{16267} erythroleukemia, M6
{23836} erythroleukemia, M6
{23806} erythroleukemia, M6
{16273} erythroleukemia, M6, PAS-positive chunks
{16274} megakaryocytic blasts, M7. See the platelets budding?
{23812} megakaryocytic blasts, M7
{23821} megakaryocytic blasts, M7
{23818} megakaryocytic blasts, PAS-positive, M7
{46340} gingival involvement; this is common in M4 & M5

Untreated AML is fatal in a few weeks. Today, many AML patients have prolonged survival and probably cures.
"Adverse prognosis" features -5 / del(5q), -7 / del(7q), -17, -18, inv(3)/t(3;3), t(6;9), t(9;22) and/or complicated cytogenetics (i.e., three or more unrelated abnormalities).
The others, including those with no chromosomal abnormalities, are "intermediate prognosis."
Acute promyelocytic leukemia has the best response to treatment, with 98% adjusted six-year survival (Blood 110: 59, 2007).
A solid growth of myeloblasts is a CHLOROMA or GRANULOCYTIC SARCOMA or NONLEUKEMIC MYELOID SARCOMA. This most malignant of solid tumors turns green (Greek "chloros", as "chlorine" or "chlorophyll") on exposure to air (why?) It's not common, but is treated as, and responds similarly to, AML, with a somewhat better chance of cure (Cancer 113: 1370, 2008).
TRANSIENT MYELOPROLIFERATIVE DISORDER ("transient leukemia") is seen in the first weeks of life in around 10% of children with Down's trisomy 21. The white count rises very high with many blasts (tending to be knobby, like megakaryoblasts, or else to resemble minimally differentiated AML), and platelets are likely to be high. There are usually not many blasts in the marrow, but there's a tendency to involve the liver. It usually self-cures, but these children are at risk for later acute myeloid / megakaryocytic leukemia (signature mutation in both conditions in GATA1: Lancet 361: 1617, 2003; Blood 101: 4301, 2003; Blood 102: 2960, 2003; J. Ped. 687, 2006; Blood 111: 2991, 2008); the occasional child without Down's who gets transient myeloproliferative disorder in the nursery is at similar risk.
THE MYELODYSPLASTIC SYNDROMES ("THE PRELEUKEMIAS", Carl Sagan's disease): Mayo Clin. Proc. 70: 673, 1995; Am. J. Clin. Path. 119(S1): S-58, 2003.
This is a family of disorders in which there are problems in producing red cells, granulocytes, and platelets. What has happened is that the normal precursor cell that gives rise to all three has been replaced by a mutant clone.
* A 5q- predicts a good response to the thalidomide analogue lenalodomide (NEJM 352: 549, 2005). * Isochromosome 17q, still mysterious, is a common lone abnormality (Blood 94: 225, 1999); my late mother, who received radiation as a child for osteomyelitis, had Ph'-negative CML with just this signature; this is also a common known hit in CML's progression to blast crisis.
Unlike most of the other neoplasms of white cells, several genes are known in which point mutations confer an unfavorable prognosis (NEJM 364: 2496, 2011).

* Subclassification is useful. The old FAB classification:
1. Refractory anemia (poor hemoglobinization, too few red cells)
2. Refractory anemia with ringed sideroblasts (>15% of nucleated red cells)
3. Refractory anemia with excess blasts (5-20% myeloblasts)
4. Refractory anemia with excess blasts in transformation (20-30% myeloblasts)
5. Chronic myelocytic leukemia
Ask your hematologist what this all means. The World Health Organization put out a new, more-elaborate clasification with criteria in 2002 (Blood 100: 2292, 2002) and tweaked it in 2008 with instructions on how to distinguish cell types; leave it to us.
Most patients are older adults, who sometimes are symptomatic. In the more aggressive forms, death follows in a few years. Often these patients are asymptomatic, and the problem is detected on routine screening.
As you would expect, this disease pattern has a propensity to transform into acute myeloid leukemia, and does so in a large minority of cases.
![]()
|
|
![]() Myelodysplastic syndrome
Myelodysplastic syndrome
Odd megakaryocyte / giant platelet
AFIP
CHRONIC MYELOID LEUKEMIA ("chronic myelogenous leukemia", "well-differentiated granulocytic leukemia"): all about it Lancet 370: 1127, 2007
This is cancer of the myeloid stem cells in which there is overgrowth of normally-maturing myeloid cells
Radiation and exposure to chemicals (notably benzene) are known risk factors. Most of the time, the disease seems to strike at random.
Patients typically have high counts of neutrophils and their precursors (and almost always basophils). These are normal (functionally and morphologically) for all intents and purposes, except that for some reason they lack cytoplasmic alkaline phosphatase.
Preposterously high white counts (>100,000 or so) are likely to result in white cells plugging important small vessels ("leukostatic ischemia" of the brain, etc.)
While the spleen is likely to be enlarged in all the common leukemias, chronic myeloid leukemia typically produces huge spleens (down almost to the pubic hair). There will usually be some little infarcts.
Occasional CML cases have predominance of basophils (itchy) or eosinophils. Serum vitamin B12 is likely to be elevated due to elevation of its binding protein; this can happen in other myeloproliferative disorders.
{10769} CML, splenomegaly
{10763} CML, peripheral blood
{12359} CML
{23863} CML (note the basophil)
{23866} CML,
leukocyte
alkaline phosphatase stain (black; note the cells are not stained black)
|
|
Both granulocytic series and "benign" monocytes and erythroid precursors bear the distinctive Philadelphia chromosome, a translocation between chromosomes 9 and 22. This produces a new gene (bcr/c-abl) which is a potent oncogene. Even "Philadelphia negative" cases have this new gene.
{12371} Philadelphia chromosome
![]() Chronic granulocytic leukemia
Chronic granulocytic leukemia
Philadelphia chromosome
WebPath Photo
The disease eventuates, after a few years, in BLAST CRISIS, with or without (50%/50%) a previous accelerated phase. Blasts appear in the circulation in large numbers (30% or more), and death follows quickly as they overwhelm the marrow and body. This is not very treatable.
{23869} CML, blast crisis
{12365} CML, blast crisis
* Because leukemias by their nature are spread around the body, there is no "staging". In CML, you may hear of three "phases" (early, accelerated, blast crisis).
Traditional chemotherapy with busulfan or hydroxyurea controls symptoms during the chronic phase but neither speeds nor delays blast crisis.
Newer therapies (alpha-interferon with or without cytarabine) suppress the leukemic clone and do prolong survival.
Historically, the only hope for a cure was allogenic bone marrow transplantation. Watch the outcome of people treated long-term with the new biotech products.
In around 30% of these cases, the blasts express lymphoid differentiation (TdT, etc.) T-cell blast crisis: Am. J. Cln. Path. 107: 168, 1997.
WARNING: The following "myeloproliferative diseases" are all "tumors of the multipotent myeloid stem cell", and can transform into one another (usually from a mild one to a bad one):
polycythemia vera rubra
hemorrhagic ("essential") thrombocythemia
primary myelofibrosis
idiopathic "aplastic anemia"
chronic myelogenous leukemia
In the 1990's, it became clear that most cases of aplastic anemia are caused by T-cell-mediated attacks on the hematopoietic marrow. This explains why...
A key to the autoimmunity in aplastic anemia may have been discovered. Many of these people have a clone of T-cells with mutated perforin (Blood 109: 5234, 2007), the same locus that's mutated in familial hemophagocytic lymphohistiocytosis.
Other cases of acquired aplastic anemia seem to be the result of running out of telomere length during aging. Some families and some individuals have less telomerase than others (to oversimplify, but the impact seems real): NEJM 352: 1413, 2005.
Once uniformly fatal, the disease is now often controllable using marrow / stem cell transplantation and/or immunosuppression (Blood 108: 2509, 2006).
CHRONIC LYMPHOCYTIC LEUKEMIA ("CLL", "well-differentiated lymphocytic leukemia"; "the liquid phase of well-differentiated lymphocytic lymphoma", etc.) Lancet 371: 1017, 2008.
|
|
|
|
|
This indolent cancer is a clone of B-cells that multiply slowly and do nothing useful. The diagnosis is made by finding a count of 5000 or more lymphocytes of appropriate phenotype circulating in the blood.
If the cells have nucleoli, it's more likely to be called B-cell prolymphocytic leukemia and to behave more aggressively.
Often you can find growth centers (where the cells are slightly larger and perhaps show nucleoli) in solid-phase well-differentiated lymphocytic lymphoma; these have the same molecular markers as classic CLL.
"T-cell CLL", with a peripheral smear looking like CLL, is now renamed "T-cell prolymphocytic leukemia", an uncommon and aggressive disease. There are of course T-cell receptor rearrangements, and most often inv 14(q11;q32).
By contrast, "monoclonal B-cell lymphocytosis" (Am. J. Clin. Path. 139: 390, 2013) is utterly harmless and (we now know) extremely common; it's a clone of B-cells in the blood (never more than 5000).
The one known risk factor is ataxia-telangiectasia (homozygotes, very likely heterozygotes), and not surprisingly, this gene is often mutated in sporadic cases (Lancet 353: 26, 1999.)
* Nowadays it is commonplace to check for a mutation in IgVH (immunoglobulin variable region); if mutated, the course is supposed to be more indolent.
* CLL has a few other common genetic markers but they also occur in other B-cell neoplasms (Semin. Hem. 36: 171, 1999.
The disease is often an incidental finding, when a CBC shows preposterously a high lymphocyte count.
{08784} CLL
{12389} CLL
{12404} CLL going bad (some blasts)
{12386} CLL with smudges
Paraneoplastic syndromes are more troublesome in this disease than in most other leukemias.
Around 15% of patients get autoimmune hemolytic anemia.
The lymphocytes do somewhat suppress the heathy plasma cells, and the patients have troubles with infections.
* A few percent develop a marker paraprotein, usually kappa IgM, or get mu heavy chain disease.
Patients with anemia or thrombocytopenia from CLL survive around 2 years. Asymptomatic people with CLL as an incidental finding generally survive more than ten years.
A few percent of CLL / SLL patients develop a diffuse large-cell lymphoma. This is the rapidly-fatal RICHTER'S SYNDROME (NEJM 324: 1267, 1991); predisposing factors remain mysterious (Cancer 67: 997, 1991).
* Around 1% of CLL terminates as ALL ("blast crisis of CLL").
The treatment of troublesome CLL has been revolutionized by the introduction of the oral Bruton tyrosine kinase inhibitor ibrutinib (NEJM 369: 32, 2013).
* PROLYMPHOCYTIC LEUKEMIA is an uncommon, aggressive variant of CLL.
Chemotherapy for end-stage CLL: NEJM 330: 319, 1994. No miracles.
* It will not surprise you to learn that many people have "pre-CLL", or "monoclonal B-cell lymphocytosis", detectable only by zealous search for the mutated cells by pathologists. Each year there's only about 1% chance of transformation (NEJM 359: 638, 2008; don't look for it Blood 119: 4358, 2012).
HAIRY CELL LEUKEMIA (Mayo Clin. Proc. 87: 67, 2012)
This distinctive leukemia is named for the many hair-like projections on its surface.
It was of unknown histogenesis (* confusing surface markers) until its molecular genetics and antigenic markers established it as a B-cell neoplasm" (Blood 104: 250, 2004; Am. J. Clin. Path. 125: 251, 2006).
Patients have circulating "hairy lymphocytes"; usually have big spleens and are sometimes anemic, neutropenic, and/or thrombocytopenic. Treatment is often unnecessary or can be delayed until the disease gets symptomatic. Bone marrow aspiration is likely to be unsuccessful (dry tap) "because the cellular hairs tangle with one another" (* more likely, because TGF-beta1 from the tumor induces extra reticulin fibrosis: J. Clin. Inv. 113: 676, 2004.
The hairs are quite distinctive, and the diagnosis is clinched by the finding of TARTRATE-RESISTANT ACID PHOSPHATASE (TRAP) in these cells.
The genetics of hairy cell leukemia are finally being discovered. A trademark (probably driver) BRAF mutation (to the familiar BRAF V600E known from other tumors) has just been found in each of 48 hairy-cell patients (NEJM 364: 2305, 2011; assays Blood 119: 3151, 2012; Am. J. Clin. Path. 138: 153, 2012; stain Am. J. Surg. Path. 36: 1796, 2012). For refractory hairy-cell leukemia, therapy targeting BRAF with vemurafenib seems effective.
* Staining for CD103 for hairy cell -- Am. J. Clin. Path. 139: 220, 2013.
During the 20th century, the only treatment for this disease was splenectomy, which helped. Today, most patients get a lasting remission after taking a course of cladribine (2-chlorodeoxyadenosine, 2-CdA) or pentostatin (deoxycoformycin, a purine analogue that's a naturally-occurring antibiotic). Update Cancer 104: 2442, 2005; Blood 109: 3672, 2007. These can be repeated is required if the disease recurs (which it often doesn't), and there are additional treatments that give results if the disease becomes resistant.
* There is a variant that features mutated p53 instead of BRAF V600# and is much more common in men and is harder to treat. There's also a Japanese variant that responds very well to cladribine.
{23872} hairy cell leukemia
{10766} hairy cell leukemia, spleen (top; normal at bottom)
{16543} hairy cell leukemia, TRAP stain (red)
{23875} hairy cell leukemia, TRAP stain (red)
{13925} hairy cell leukemia, TRAP stain (red)
{16541} hairy cell leukemia, TRAP stain (red)
{23881} hairy cell leukemia, bone marrow biopsy (trust me)
{42117} big spleen in hairy cell leukemia, foot ruler
POLYCYTHEMIA VERA ("Osler's polycythemia", "P. V. rubra", etc.; Mayo Clin. Proc. 78: 174, 2003; Arch. Path. Lab. Med. 130: 1126, 2006)
By convention, POLYCYTHEMIA (a better synonym is ERYTHROCYTOSIS) describes an abnormally high hemoglobin. Classification:
ABSOLUTE POLYCYTHEMIA (i.e., increased circulating red cell mass):
PRIMARY POLYCYTHEMIA (i.e., the main problem is with the red cells)
Polycythemia vera rubra
NOTE: Cancer of the normoblasts (i.e., AML-M6) isn't considered a polycythemia
SECONDARY POLYCYTHEMIA (i.e., the main problem is elsewhere)
Effective renal arterial hypoxia
Emphysema
Sleep apnea
Tetralogy of Fallot
Hemoglobins with too much oxygen affinity
Etc., etc.
For a first-person story of injectable bioengineered erythropoietin and bicycle racing, including how athletes beat the tests by infusing huge amounts of normal saline by vein moments before having their hematocrits checked, see Sci. Am. 298(4): 82, 2008.
Genetic errors in erythropoietin production or sensitivity ("familial erythrocytosis", for example HIF2A: NEJM 358: 162, 2008)
Erythropoietin-producing tumors
Renal cell carcinoma
* Hepatocellular carcinoma
* Cerebellar hemangioblastoma (?!)
Anabolic steroid users
After kidney transplant (over-zealous proximal tubule produces erythropoietin)
Altitude (above about 10,000 feet for a long time? Expect problems. Stroke risk: Stroke 26: 562, 1995. Pulmonary vein thrombosis: Hum. Path. 21: 601, 1990.
"Primary familial polycythemia" / "congenital primary erythrocytosis" (truncated erythropoietin receptor stuck in the "on" position; has produced a family of elite athletes; (J. Clin. Invest. 102: 124, 1999; others Proc. Nat. Acad. Sci. 103: 654, 2006)
* Erythropoietin-dependent polycythemias (altitude, post-transplant) can be ameliorated using ACE inhibitors, which is puzzling: Lancet 359: 663, 2002.
RELATIVE POLYCYTHEMIA (i.e., dehydration)
Polycythemia vera is a proliferation of stem cells (again, the common precursors of red cells, granulocytes, and megakaryocytes). This time, they are very erythropoietin-sensitive and mostly mature into red cells.
The cells are the common ancestors of red cells, neutrophils, and megakaryocytes. Over the course of years, these stem cells replace the normal stem cells of the marrow. Their progeny, however, are fully functional. (Neutrophils even have normal alkaline phosphatase levels.)
In addition to a high red cell count, white cells and platelets are likely to be high.
On biopsy, expect to see a very hypercellular marrow, with all cell lineages increased. In the late stages, there is often marrow fibrosis ("burned out PVR", "postpolycythemic phase", * PDGF effect?) or replacement by blasts (transformation to acute myelogenous leukemia -- still no good treatment Cancer 104: 1032, 2005).
The trademark mutation in JAK2 (Janus kinase; NEJM 356: 444, 2007) that is usually present (* JAK2V617F) is now famous. Mouse with the mutation -- Blood 120: 166, 2012.
The new JAK2 inhibitors are now widely used. For example, ruxolitinib seems to be superior to standard treatment for polycythemia vera (NEJM 372: 426, 2015).
This is a disease of older middle-age. Until the last stages, patients are troubled primarily by the increased volume of hyperviscous blood.
This causes congestion of most organs ("the plethoric face", etc.)
More troubling, the stasis of gooey blood in the veins promotes clotting.
Or distended veins can rupture (GI bleeding, hemorrhagic stroke). Eventually these patients get platelet problems, too, which does not help the bleeding tendency.
Minor mystery of medicine: Itching after taking a hot shower is very suggestive of PVR.
The mainstay of treatment for polycythemia vera is regular phlebotomy, to keep the red cell count down.
These patients' survival curves are nearly as good as normal folks. For all three of the JAK2 diseases, the major killer is the tendency of the disease to turn into acute myelogenous leukemia.
The once-popular practice of giving these patients radioactive phosphorus resulted in a greatly increased rate of transformation to acute leukemia, turning a not-so-bad, easy-to-control disease into a lethal, untreatable one. Later, the same thing happened with trials of chemotherapy.
The traditional criteria for the diagnosis of PVR:
A1... Increased RBC mass (>=36 mL/kg; >=32 mL/kg; the math comes to a hematocrit of 52 for a man, 46 for a woman)
A2... Normal arterial PO2
A3... Splenomegaly
B1... Platelets greater than 400,000/L
B2... WBC >=12,000/L
B3... Leukocyte alkaline phosphatase over 100 in the absence of evidence of infection
B4... Elevated serum vitamin B12.
Make the diagnosis if:
(1) You have A1 + A2 + A3, or
(2) A1 + A2 + any two B's.
In 2007, the World Health Organization suggested requiring two major criteria (hemoglobin >18.5 g/dL;for men, >16.5 g/dL for women, plus a functionally active JAK2 mutation. Minor criteria are one of these -- characteristic bone marrow morphology, too-low serum erythropoietin, and the ability of red cell colonies to grow in tissue culture without erythropoietin.
* Thrombosis, the most troublesome aspect of this disease, seems to be much less of a problem if patients are simply given low-dose aspirin (NEJM 350: 114, 2004).
* Keeping the hematocrit below 45% (rather than just below 50%) seems to be good for preventing cardiovascular events (NEJM 368: 22, 2013).
![]() Polycythemia
Polycythemia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
PRIMARY MYELOFIBROSIS ("myelofibrosis with myeloid metaplasia"; "agnogenic myeloid metaplasia"; "myelosclerosis"); JAMA 303: 2513, 2010; Mayo Clin. Proc. 87: 25, 2012
Primary myelofibrosis is a proliferation of neoplastic stem cells in the bone marrow (which merely becomes hypercellular) and the red pulp of the spleen (which enlarges greatly). For some reason, the marrow tends to develop increased reticulin / collagenize, "burn out" and become fibrotic.
* The WHO criteria require (1) megakaryocyte proliferation and atypia and usually fibrosis of the marrow; (2) not being one of the other recognizable myeloprolifeative diseases; (3) some clonal marker such as JAK2 or one of the others or at least not chronic inflammation or cancer; (4) Two of the following: leukoerythroblastic smear; increased serum LDH; anemia; palpable spleen. These are going to be discarded when the molecular signatures are better-defined.
Historically, ionizing radiation and benzene exposure are known risk factors.
We know it's tumor since there's a group of common translocations; however, which ones are present doesn't impact prognosis at least much (Cancer 107: 2801, 2006). Most now seem to have mutated JAK2, and it seems likely this will soon define the disease.
As in polycythemia vera, the cells that enter the blood are fully functional. This time, there is no tendency to over-produce red cells; neutrophils may be super-abundant and "left-shifted" (Philadelphia-chromosome negative, of course), or there may be neutropenia, or the WBC may be normal. Platelets are unaffected or even increased (until maybe very late).
This is a disease of older adults. Patients are most likely to be troubled by feeling full after they've eaten just a little (why?).
Examine the peripheral smear. Red cells made in the spleen tend to be teardrop-shaped (one form of "poikilocyte"), and nucleated red-cell precursors from the spleen are more likely to escape into the circulating blood. Leukocyte alkaline phosphatase is likely to be high.
* The year 2010 saw the first medication effective for primary myelofibrosis, an inhibitor of JAK1/JAK2, whether or not the latter bears the trademark mutation (NEJM 363: 1117, 2010). Ruxolitinib, a JAK1 and JAK2 inhibitor for myelofibrosis: NEJM 366: 787 & 799, 2012).
{12302} teardrop reds
NOTE: The important term LEUKOERYTHROBLASTIC SMEAR refers to the presence in the bloodstream of young red cells and immature granulocytes. You'll see this when they are "being pushed out of their place of origin too fast" by distortion of the marrow environment:
bone marrow infiltration
primary myelofibrosis
metastatic carcinoma
lymphoma
leukemia
bone marrow hyperplasia / extramedullary hematopoiesis
severe hemolysis, etc.
After this process has been underway for several years, the bone marrow undergoes dense fibrosis. Long mysterious, it is now clear that the marrow fibroblasts are responding to over-production of PDGF (platelet-derived growth factor) and * transforming growth factor-beta produced by abnormal megakaryocytic cells.
{13799} myelofibrosis, marrow core biopsy
{24788} myelofibrosis, marrow core biopsy
{13802} myelofibrosis, reticulin stain
Patients with "myeloid metaplasia / myelofibrosis" ultimately die of cytopenia or transformation to acute leukemia. Not surprisingly, those that eventually transform into acute leukemia tend to have a few circulating blasts at diagnosis (Cancer 112: 2726, 2008).
The old term "agnogenic" means "of unknown cause" (i.e., it's a synonym for "idiopathic"; compare "agnostic").
* Diagnosticians: Unexplained myelofibrosis WITHOUT splenomegaly suggests M7 AML, burning-out CML, or burned-out polycythemia vera; look also for carcinoma cells.
* Autoimmune myelofibrosis may result from lupus or "just happen"; future pathologists recognize it by the absence of any abnormalities of the remaining marrow cells, and clusters of lymphocytes. Am. J. Clin. Path. 116: 211, 2001.
PLASMA CELL MYELOMA ("multiple myeloma", "malignant plasmacytoma") NEJM 336: 1657, 1997; Lancet 363: 875, 2004; for pathologists dealing with the difficult diagnostic cases Am. J. Clin. Path. 136: 168, 2011 (let us worry about them).
This is cancer of the plasma cells (i.e., cancer of B-cells that are differentiated enough to secrete an immunoglobulin and/or a light chain (kappa or lambda, though of course never both), or at least to look like plasma cells).
Myeloma is only slightly less common than leukemia or lymphoma. The typical patient is in his or her fifties.
The etiology is obscure, and the disease seems to strike at random.
* A majority of patients are men. Black people have a slightly increased rate over people of other ancestry. Like lupus and sarcoidosis or sickle cell, nobody's going to base the diagnosis (or hopefully anything else) on one's race.
The term "multiple myeloma" comes from its tendency to make multiple holes ("lytic lesions") in the bone marrow ("myelo-") and nearby cortex. The effect is mediated, at least in part, by lymphotoxin (TNF-beta). Cancer of plasma cells always involves bone, but only about half of cases feature real "punched-out" x-ray lesions. The remaining patients have diffuse disease and suffer precocious osteoporosis. I have never used the term "multiple myeloma" and urged others not to do so either, and it finally (2008) seems to be going out of use.
* Future clinicians / hardcore pathology students: Here are your
CRITERIA FOR THE DIAGNOSIS OF PLASMA CELL MYELOMA! 2009 version
A serum or urine paraprotein (unless it's true non-secretory myeloma) and
One of these signs of end-organ damage: Hypercalcemia, myeloma kidney, anemia, bone lytic lesions
* INDOLENT MYELOMA: Old diagnosis. More than 30% bone marrow plasma cells, IgG spike <7 gm/dL or IgA spike <s;5 gm/dL; fewer than three lytic lesions; no anemia, hypercalcemia, or renal involvement
SMOLDERING MYELOMA: 10% or more plasma cells in the marrow, paraprotein 3 gm/dL or more; no bone lesions, anemia, hypercalcemia, or renal involvement (i.e.,
nothing's damaged)
MONOCLONAL GAMMOPATHY OF UNCERTAIN SIGNIFICANCE: <10% plasma cells in the marrow
(but who wants to check?); spike less than 3 gm/dL; no lytic bone lesions no anemia, hypercalcemia, or renal involvement
Clonal plasma cells, 10% or more in the marrow, or any percent elsewhere AND
PLASMA CELL MYELOMA: As above.
{08462} bony lesions of myeloma (skull and spine)
{27327} bony lesions of myeloma (skull)
{13769} skull lesions of myeloma
{10760} skull lesions of myeloma
{10754} bone lesions of myeloma
{10757} osteoporosis of myeloma
{46197} femur lesions in myeloma
{46198} rib lesions in myeloma
{27329} spike, probably monoclonal gammopathy of uncertain significance, since normal albumin and
gamma seem not to be suppressed

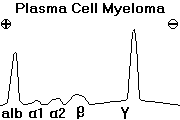
The monoclonal protein (immunoglobulin or chain) produced by an abnormal clone of multiple myeloma cells is called the M-PROTEIN.
If there's a complete antibody, you'll see it on serum protein electrophoresis.
Free light chains may be produced along with, or instead of, a complete immunoglobulin. They pass easily through the glomerular basement membrane, so you will probably not find them in the bloodstream if the kidneys are working. Instead, they accumulate in the urine, where they are called BENCE-JONES PROTEIN. About 2/3 of myeloma patients produce Bence-Jones protein; a majority of these also produce some complete immunoglobulin.
Later on, Bence-Jones protein plugs up the renal tubules, and contributes to "myeloma kidney", which we'll study in the "renal pathology" section.
You remember that plasma cell myeloma is an important cause of amyloidosis AL, which doesn't help renal function, either.
Here's a breakdown on types of M-proteins:
55%...IgG
25%...IgA
1%... IgE, IgD, or IgM monomer (* IgM myeloma Am. J. Clin. Path. 140: 519, 2013).
18%... Bence-Jones protein only
1%... no M-protein.
A PARAPROTEIN is an abundant, useless, monoclonal protein in the bloodstream. All M-proteins are paraproteins; you'll meet others. Lots of an M-protein will produce rouleaux formation; we'll talk more about this in "Clinical Pathology".
NOTE: As a rule, plasma cell myeloma does not make IgM pentamers. Waldenstrom's does this, and you won't see the typical bone changes.
To make the diagnosis, you will want to find an overabundance (>15% or so) or sheets of plasma cells (typical or weird-looking) on bone marrow.
{16554} plasma cell myeloma, cells
{16556} plasma cell myeloma, cells
{13772} plasma cell myeloma, marrow aspirate
{27330} plasma cell myeloma, marrow aspirate
{13775} plasma cell myeloma, bone marrow section
{10751} * "grape cell"
{42054} * "flame cell" (named for its staining properties)
Normally, only around 3% of bone marrow cells are plasma cells, but whenever there is widespread B-cell activation, their number can increase substantially.
* Future pathologists: In reactive plasmacytic disorders, plasma cells encircle the vessels. In plasma cell myeloma, you'll probably find plasma cells encircling fat cells.
Patients are now often getting apparent cures. Paraneoplastic problems are the greatest problem in plasma cell myeloma. Be alert for:
{17273} myeloma kidney, Bence-Jones casts with foreign body reaction
{17274} myeloma kidney, Bence-Jones casts with foreign body reaction
The tumor generally excites no fibrous or osteoblastic response. At autopsy, the tumor masses (if distinguishable) look and feel like reddish-gray jelly.
Prognosis is much better, nowadays due both to chemotherapy and to bisphosphonate management of bone disease. The ongoing "total therapy" studies is reporting prolonged remissions (cures?) in many patients (updates Blood 112: 3115, 2008; Cancer 133: 355, 2008; Cancer 112: 2720, 2008). The monoclonal bortezomib (proteasome inhibitor) is very promising (updates Cancer 110: 1042, 2007; Cancer 112: 1529, 2008). Thalidomide for refractory myeloma NEJM 341: 1565, 1999. This is now mainstream.
OTHER PLASMA-CELL PROBLEMS ("plasma cell dyscrasias", an archaic term that has hung on for some reason)
There are a variety of other MONOCLONAL PLASMA CELL PROLIFERATIONS.
We already looked at WALDENSTROM'S MACROGLOBULINEMIA and the HEAVY-CHAIN DISEASES under "non-Hodgkin's lymphomas". These are cancers of small lymphocytes with "plasmacytoid" features.
SOLITARY PLASMACYTOMAS may appear benign grossly and microscopically, and they may or may not produce immunoglobulins.
Those in bone almost always recur as plasma cell myeloma.
Those in extra-osseous sites ("plasmacytic lymphoma") may often be resected for cure.

MONOCLONAL GAMMOPATHY OF UNCERTAIN SIGNIFICANCE ("MGUS", the old "benign monoclonal gammopathy") affects maybe 3-5% of older adults (prevalence NEJM 354: 1362, 2006; old texts are wrong to suggest it is less common than plasma cell myeloma).
This is best considered a benign, disseminated proliferation of plasma cells with some potential to transform into malignancy.
The tumor cells produce a single, complete immunoglobulin (usually IgG) that may be detected on serum protein electrophoresis.
Maybe 1/4 of these people eventually go on to get sick from plasma cell myeloma, amyloidosis AL, or macroglobulinemia (Mayo Clin. Proc. 68: 26, 1993); newer work gives the rate at about 1%/year (NEJM 346: 564, 2002)
AMYLOIDOSIS B may arise in this setting, and probably all non-cancer-related amyloidosis AL cases have a hyperactive clone of plasma cells.
Smoldering myeloma (NEJM 356: 2582, 2007) features 10% or more plasma cells in the marrow, and an M-protein of myeloma proportions, but no signs of end-organ damage. It often, but not always, proceeds to myeloma over the years. See below.
 MGUS
MGUS
Pittsburgh Pathology Cases
* POEMS may arise: polyneuropathy, organomegaly, endocrinopathy (thyroid/gonads), monoclonal gammopathy (usually IgA-lambda), and skin changes. Some folks have myeloma; other just probably have an odd clone of plasma cells. The molecular etiology remains elusive (Am. J. Resp. Crit. Care Med. 157: 907, 1998 -- still not much more).
* CRYOGLOBULINEMIA TYPE I is a monoclonal immunoglobulin with poor solubility. More about this in "Clinical Pathology"!
Follow these people up for decades, and around one in four will get some kind of serious gammopathy (Mayo Clin. Proc. 68: 26, 1993).

POLYCLONAL ACTIVATION OF PLASMA CELLS ("polyclonal gammopathy") is a common finding in clinical medicine. Situations worth remembering:
THE LANGERHANS CELL HISTIOCYTOSIS FAMILY ("LCH", "Histiocytosis X", "disseminated histiocytosis"; * R&F "differentiated histiocytosis" is a typo); review for clinicians J. Ped. 127: 1, 1995; Cancer 85: 2278, 1999.
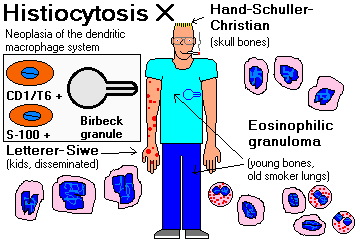
A group (probably a continuum) of lesions that are probably honest-to-goodness tumors of Langerhans-type histiocytes, a class of dendritic macrophages.
Langerhans cells in health and disease are characterized by intracellular BIRBECK GRANULES ("histiocytosis X bodies"), pentalaminar tennis-racket shaped structures of unknown significance.
{09095} Birbeck granules
{09097} Birbeck granules
![]() Histiocytosis X
Histiocytosis X
Pittsburgh Illustrated Case
![]() Histiocytosis X with Birbeck granules
Histiocytosis X with Birbeck granules
Lung pathology series
Dr. Warnock's Collection
![]() Eosinophilic granuloma of the lung
Eosinophilic granuloma of the lung
Lung pathology series
Dr. Warnock's Collection
In tissue, you will probably see a range of cells from "blasts" to well-differentiated Langerhans cells.
The former claim that histiocytosis X is "polyclonal" probably resulted from confusion of the tumor cells with non-neoplastic inflammatory cells that had entered the tumor. By the mid-1990's we knew the disease was clonal, hence a real neoplasm (NEJM 331: 154, 1994; Br. Med. J. 310: 74, 1995; Lancet 344: 1717, 1994).
Future pathologists: Histiocytosis X and the dendritic macrophages from which it derives stain for CD1/T6. They also stain with S-100.
The old names are passing out of use, but you might perhaps see the syndromes:
LETTERER-SIWE DISEASE ("acute disseminated histiocytosis", "multifocal multisystem LCH") affects small children and involves most of the body's organs. These children are now often cured with elaborate chemotherapy.
{23392} Letterer-Siwe disease. Weird histiocytes ("coffee-bean nuclei, even"). Trust me.
EOSINOPHILIC GRANULOMA ("unifocall LCH"; "granuloma" is an unfortunate misnomer) causes solitary bone lesions in young people. The histiocytes have coffee-bean nuclei and are admixed with eosinophils. Modest treatment generally is curative. There is a variant that affects the lungs of smokers.
{13688} eosinophilic granuloma
{13691} eosinophilic granuloma
{09043} eosinophilic granuloma, EM, coffee-bean nucleus (left) and eosinophil (right)
HAND-SCHÜLLER-CHRISTIAN DISEASE ("multifocal unisystem LCH", affects the skull bones and perhaps -- look for diabetes insipidus, proptosis, lytic skull lesions, fever, and rash. It's intermediate between the other two in severity.
And not in the classic scheme, but recognized now thanks to improved pathology techniques, CUTANEOUS LANGERHANS CELL HISTIOCYTOSIS, a disease of infants that often self-cures (Arch. Derm. 146: 149, 2010).
{10481} Hand-Schüller-Christian disease. Weird histiocytes. Trust me.
{21779} skull in Hand-Schüller-Christian disease
* CHESTER-ERDHEIM DISEASE ("lipid granulomatosis"; "cholesterol granulomatosis") is a rare illness in which lipid-laden non-dendritic-type macrophages infiltrate the tissues. Thankfully rare, it is clonal and seems to be a neoplasm (Hum. Path. 30: 1093, 1999).
* Every man has his own ways of courting the female sex. I should not, myself, choose to do it with photographs of spleens, diseased or otherwise.
-- Agatha Christie, "The Moving Finger"
The healthy spleen weighs 50-250 gm or less. You remember that the cells right around the arteries in the white pulp are T-cells, that there are likely to be B-cell nodules, and that the Littoral cells lining the sinuses express both macrophage and endothelial markers.
The spleen almost never gets biopsied, as it is so likely to rupture.
SPLENOMEGALY must be substantial (800 gm or so) to be palpable. Causes worth remembering:
INFECTIONS
Malaria (huge spleens)
Infectious mononucleosis family (see above)
Bacterial endocarditis (don't miss this one)
Most other bad infections (NOTE: A "septic spleen" feels soft, unlike most of the other big spleens)
CONGESTION (if longstanding, becomes "fibrocongestive")
Cirrhosis
Right-sided heart failure
Splenic vein thrombosis
Sludging of red cells
Sicklers
Polycythemia vera
Waldenstrom's
Others
Others
DISEASES OF WHITE CELLS
Chronic myelogenous leukemia (huge spleens)
Primary myelofibrosis (huge spleens)
Hairy cell leukemia (very large spleens)
All the other ones
SPLENIC OVER-DESTRUCTION OF BLOOD CELLS
Hereditary spherocytosis
Hemoglobinopathies and bad thalassemia
Immune hemolytic anemia
Immune thrombocytopenic purpura
IMMUNOREACTIVE HYPERPLASIA
Lupus
Rheumatoid arthritis
Graft rejection
STORAGE DISEASES (huge spleen)
Gaucher's (very big, wadded-kleenex macrophages)
Niemann-Pick's (very big, foamy macrophages)
Hunter's
Hurler's
SARCOIDOSIS
AMYLOIDOSIS (sago, lard)
HYPERSPLENISM is said to be present when an enlarged spleen destroys normal formed elements of blood too readily. The three causes you'll probably see are: (1) cirrhosis; (2) rheumatoid arthritis (the serious "Felty's syndrome"), and (3) Gaucher's disease. It's also one cause of thrombocytopenia in some leukemia and lymphoma patients.
{00239} Gaucher's disease, spleen
{09864} Gaucher's disease, spleen
{16216} Gaucher's disease, watered-silk ("wadded kleenex") cell from spleen
The only proof that hypersplenism was the problem is that the blood counts get better when the spleen is removed.
|
|
ACCESSORY SPLEENS (one or more) are present somewhere in the abdomen in about 25% of autopsies. If you need a splenectomy for a medical disease (i.e., immune thrombocytopenic purpura, hereditary spherocytosis), you must hope that your surgeon does not overlook a large accessory spleen.
SEPTIC SPLEEN ("nonspecific acute splenitis") is typical of serious bacterial infections. Loaded with polys and abnormally soft, the old gourmet pathologists made the comparison to "tomato paste", which is very much resembles.
HYPERPLASTIC SPLEEN usually means large germinal follicles in the white pulp. Think of systemic autoimmune disease, infectious mononucleosis, graft rejection, etc., etc.
In INFECTIOUS MONONUCLEOSIS, the spleen also becomes infiltrated with activated T-cells that give a malignant appearance. The capsule is stretched and infiltrated, making it more fragile. You're unlikely to see such a spleen unless it is removed because of rupture (sports, overzealous physical exam).
* Future pathologists: Telling hyperplasias from lymphomas in the spleen is one of your toughest calls. For help, see Am. J. Clin. Path. 99: 486, 1993.
INFARCTS are common in the spleen, and may result from atheroembolization (the twisty splenic artery is the most severely affected in the body), left-sided endocarditis, or infiltrative disease.
* Necrosis in a blood-bloated spleen (typically, in sicklers) is likely to produce iron- and calcium-rich scars called GAMNA-GANDY BODIES. The "autosplenectomized" spleen of an older sickle-cell disease patient is mostly composed of such scars.
![]() Sickle cell disease
Sickle cell disease
Autosplenectomy
WebPath Photo
PRIMARY NEOPLASMS of the spleen are uncommon. Benign tumors are almost never of any importance. Any lymphoma or endothelial neoplasm can arise here. METASTASES to the spleen are expected in most leukemias and Hodgkin's and non-Hodgkin's lymphomas, but carcinomas and sarcomas very seldom grow in the spleen.
RUPTURED SPLEEN results from blows (hard if you're healthy, lighter blows suffice for those with infectious mononucleosis; remember CPR as a cause Br. Med. J. 322: 480, 2001). Intraperitoneal hemorrhage results in a trip to surgery. If a lot of pulp escapes into the peritoneal cavity, the patient may heal with hundreds of mini-spleens over the peritoneal cavity (SPLENOSIS).
You remember the difference between sections and smears, right?

"It is futile," I said,
You lie," he cried,
* I SAW A MAN PURSUING
I saw a man pursuing the horizon;
Round and round they sped.
I was disturbed at this;
I accosted the man.
"You can never -- "
And ran on.
--Stephen Crane
(1871-1900)
 * SLICE OF LIFE REVIEW: BLOOD CELLS
* SLICE OF LIFE REVIEW: BLOOD CELLS
10110 ff blood
{10766} leukemia, hairy cell and normal
{12275} anemia, iron deficiency; normal
{13715} lymphocyte, normal
{13868} red blood cell, normal blood
{13910} red blood cell, normal
{14702} polymorphonuclear leukocyte, normal
{14703} polymorphonuclear leukocyte, normal
{14704} polymorphonuclear leukocyte, normal
{14705} polymorphonuclear leukocyte, normal
{14705} polymorphonuclear leukocyte, normal
{14706} polymorphonuclear leukocyte, normal
{14707} polymorphonuclear leukocyte, normal
{14708} eosinophil, normal
{14709} eosinophil, normal
{14710} basophil, normal
{14711} basophil, normal
{14712} monocyte, normal
{14713} monocyte, normal
{14714} monocyte, normal
{14715} monocyte, normal
{14716} lymphocyte, large
{14717} lymphocyte, large
{14718} lymphocyte, normal
{14719} lymphocyte, normal
{14720} lymphocyte, normal
{14721} lymphocyte, normal
{14722} reticulocytes, normal
{14723} reticulocytes, normal
{14724} red blood cell, abnormal
{14725} red blood cell, abnormal
{14726} platelets, normal
{14727} platelets, normal
{14728} pronormoblast, normal
{14729} pronormoblast, normal
{14730} basophilic normoblast, normal
{14731} basophilic normoblast, normal
{14732} normoblast
{14733} normoblast
{14734} polymorphonuclear leukocyte & * lymphocyte
{14735} polymorphonuclear leukocyte & * lymphocyte
{14736} normoblast series
{14737} normoblast series labelled
{14738} myelocyte, normal
{14739} myelocyte, normal
{14740} * granulocyte series
{14741} * granulocyte series (labelled)
{14742} myelocyte, band form
{14743} myelocyte, band form
{14744} myelocyte, normal
{14745} myelocyte, normal
{14746} myelocyte, normal
{14747} myelocyte, normal
{14748} myelocyte, normal
{14749} myelocyte, normal
{14750} myelocyte, normal
{14751} myelocyte & megakaryocyte, normal
{14752} myelocyte & megakaryocyte, normal
{15193} plasma cell, #23
{15205} thymus, adult
{15564} thymus, normal
{15565} thymus, normal
{15566} thymus, normal
{15567} thymus, normal
{16175} red blood cell, normal
{20782} polymorphonuclear leukocyte, normal
{20783} monocyte
{20784} platelets, circulating blood
{20785} monocyte
{26230} polymorphonuclear leukocyte, normal
{40179} thymus, normal
{46538} red cell, normal
 * SLICE OF LIFE REVIEW: LYMPHOID ORGANS
* SLICE OF LIFE REVIEW: LYMPHOID ORGANS
{11750} spleen, normal
{11751} spleen, normal
{11753} lymph node, normal
{11797} spleen, normal
{11805} spleen, normal unfixed
{14753} thymus, human fetal
{14754} thymus, human fetal
{14755} thymus, juvenile
{14756} thymus, juvenile
{14757} thymus, adult
{14758} thymus, adult
{14759} thymus, juvenile
{14760} thymus, juvenile
{14761} hassall's corpuscles
{14762} hassall's corpuscles
{14763} hassall's corpuscles
{14764} hassall's corpuscles
{14765} thymus (septum)
{14766} thymus (septum)
{14767} spleen, normal
{14768} spleen, normal
{14769} spleen, pulp
{14770} spleen, pulp
{14771} spleen (trabeculae), normal
{14772} spleen (trabeculae), normal
{14773} spleen (trabecular artery), normal
{14774} spleen (germinal center), normal
{14775} spleen (germinal center), normal
{14776} spleen (venous sinus), normal
{14777} spleen (venous sinus), normal
{14778} spleen (scanning em)
{14779} spleen (scanning em)
{14780} lymph node, normal
{14781} lymph node, normal
{14782} lymph node cortex, normal
{14783} lymph node cortex, normal
{14784} lymph node, medulla
{14785} lymph node, medulla
{14786} lymph node, normal
{14787} lymph node, normal
{15189} lymph node and subcapsular sinus, #23
{15190} lymph node, primary nodule
{15191} lymph node, germinal center
{15192} lymph node, medulla
{15194} spleen, #24
{15195} spleen, * red pulp and white pulp
{15196} spleen, central artery
{15197} spleen, central artery and germinal cent
{15198} spleen, trabeculae
{15199} thymus, #25
{15200} thymus, cortex
{15201} thymus, hassall's corpuscle
{15202} thymus, medulla
{15203} thymus, epithelial reticular cell
{15568} spleen, normal
{15569} spleen, normal
{15570} spleen, normal
{15571} spleen, normal
{15769} spleen, normal
{15770} spleen, normal
{20200} spleen, normal
{20799} lymph node, overview
{20800} lymph node, cortex
{20801} lymph node, medulla
{20802} lymph node, subcapsular sinus
{20803} lymph node, secondary nodule
{20804} lymph node, primary nodule
{20805} spleen, normal histology
{20806} spleen, red pulp
{20807} spleen, white pulp
{20808} spleen, central artery
{20809} spleen, red pulp
{20810} spleen, secondary nodule
{20811} spleen, trabecula
{20812} thymus, overview
{20813} thymus, medulla
{20814} thymus, cortex
{20815} thymus, hassall's corpuscle
{20827} tonsil, palatine
{20828} tonsil, pharyngeal
{24782} lymph node, normal
{24783} lymph node, normal
{36344} lymph node, normal
{36347} lymph node, normal
{36350} lymph node, normal cytology
{36353} lymph node, normal cytology
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about diseases of the white blood cells to consult these standard textbooks.
In my notes, the most helpful current journal references are embedded in the text. Students using these during lecture strongly prefer this. And because the site is constantly being updated, numbered endnotes would be unmanageable. What's available online, and for whom, is always changing. Most public libraries will be happy to help you get an article that you need. Good luck on your own searches, and again, if there is any way in which I can help you, please contact me at scalpel_blade@yahoo.com. No texting or chat messages, please. Ordinary e-mails are welcome. Health and friendship!


| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |


 Breast / Heme (?!)
Breast / Heme (?!)

