Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
verify here. |
Cyberfriends: The help you're looking for is probably here.
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dysfunctional ribosomes (?!)
ENOUGH NORMOBLASTS NOT BEING MADE
Spherocytosis: Spherocytes (look smallish as they don't lie flat, no central pallor) on peripheral smear; Increased MCHC (hyperchromasia -- remember spherocytosis as the one genuine cause; potassium leaks out of the damaged lipid bilayer and takes water with it)
Paroxysmal nocturnal hemoglobinuria: New molecular assays have replaced the old Ham test (acid hemolysis)
Enzyme deficiencies: Heinz bodies
Abnormal hemoglobins: Target cells ("codocytes"; Mexican hat cells; if it's extremely humid or the tech uses a blow-dryer on the slide, you'll get these as artifact)
Sickle cell disease: Sickled cells (drepanocytes; if in doubt, remember sickled red cells have no central pallor)
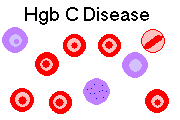
Hemoglobin C disease (two doses): Hemoglobin C crystals (add a drop of vinegar to see them better; it also helps if the spleen is gone)
Hemoglobin SC disease: Hemoglobin SC ("birds in flight") crystals
Immune hemolysis: Spherocytes (especially true microscopherocytes, because portions of the membrane have been removed); Positive direct Coombs' test. (Savvy labs now give you a free Coombs test if you're found to have unexplained elevated reticulocytes: Am. J. Clin. Path. 124: 129, 2005).
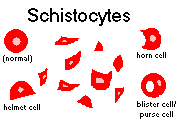
Some mechanical hemolysis (clostridia, burns, prosthetic heart valves, microangiopathy): Schistocytes and other fragments on peripheral smear
Acute hemorrhage: Increased reticulocytes (it usually takes about five days for them to appear. You remember about CFU-E's, erythropoietin and all that; there's generally a thrombocytosis during recovery from a bad bleed)
Enough hemoglobin not being made: Decreased MCV (microcytosis); Decreased MCHC (hypochromia)
Not enough usable iron: Decreased serum iron
Actual iron deficiency (none stored): Very low serum ferritin (reliable); Zero stainable marrow iron stores (but who needs to check?); low serum iron (usually), increased total iron binding capacity / low transferrin saturation (less reliable than serum ferritin).
Free erythrocyte protoporphyrin: Screening tool for iron deficiency, best in kids. Why? (Also picks up lead poisoning. Why?)
Anemia of inflammation / sideroblastic anemias: Increased serum ferritin; total iron binding capacity not indreased
Sideroblastic anemia: Dimorphic population (some forms); Ringed sideroblasts in marrow
Lead poisoning: Coarse basophilic stippling (maybe -- lead inhibits ribonuclease)
Thalassemias: Coarse basophilic stippling (maybe); Pancake cells ("leptocytes")
Enough normoblasts not being made: Decreased reticulocytes
Not enough nucleic acid being made: Increased MCV (macrocytosis); Hypersegmented PMN's
B12 deficiency: Low serum B12; Perform a Schilling test (if you can get the isotope)
Classic pernicious anemia: Schilling test becomes normal on addition of extrinsic factor; with the isotope unavailable, just check the blood for antibodies against gastric parietal cell antibodies (maybe 70% sensitive and 50% specific if you're just fishing) and maybe intrinsic factor antibodies (J. Clin. Path. 62: 439, 2009, even less sensitive and rare if anti-parietal cell antibodies are not present); do a good H&P, make a presumptive diagnosis and treat
Folic acid deficiency: Low red cell folate (avoids the pitfall of plasma fluctuations with diet)
Infiltrative disease of bone marrow: Teardrop red cells (it's still unsettled, but your lecturer believes that teardrop red cells are the ones made in spleen; see also Am. J. Heme 21: 415, 1986.)
Note: In alcohol abuse and serious liver disease, RBC's supposedly become a bit macrocytic from their membranes taking up extra lipids. This is still under study.
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- RBC / Bleeding
Lectures follow Textbook
QUIZBANK
Hematology Lab
Blood Bank (if and only if we have a chance to cover transfusion medicine in this unit)
Blood & Lymph #'s 1-132, 140-177
INTRODUCTION

Anemia affects at least a quarter of the world's population, with a disproportionate burden among children and non-pregnant women in the middle- and low-income nations (Lancet 378: 2123, 2011) -- of course the major causes are dietary iron deficiency, dietary folate and B12 deficiency, hookworm, schistosomiasis, and malaria. The loss of productivity and human potential is terrible.
Workup of anemia in adults: Mayo Clin. Proc. 78: 1274, 2003. In the newborn: Ped. Clin. N.A. 51: 1087, 2004. What to do generally if there's a CBC abnormality: Mayo Clin. Proc. 80: 923, 2005.
Review the following terms:
Hemoglobin
Hematocrit
Red cell indices (mean corpuscular volume, mean corpuscular hemoglobin concentration, mean corpuscular hemoglobin)
RDW: Red cell size distribution width. The coefficient of variation of the red cell volume. The measure of anisocytosis (cell size variability).
Do you remember all those hemoglobins?
Hemoglobin is four globin chains plus four heme units (porphyrin plus iron):
2α + 2β = Hgb A
(α2 β2A) 96% in normal adults
2α + 2 δ = Hgb A2
(α2 δ2) 3% in normal adults
2α + 2 γ = Hgb F
(α2 γ2) 1% in normal adults
(Hgb F is the main hemoglobin before birth, and remains abundant until the child is two years old.)
Do you remember the hemoglobins seen only in disease?
Wrong ways of combining normal chains:
4β = Hgb H (β4)
4 γ = Hgb Bart's (γ4)
Mutations:
2α + 2βS = Hgb S (α2βS2)
2α + 2βC = Hgb C (α2βC2)
2α + 2 β-δ Lepore = Hgb Lepore(α2 β-δ2, which is not made in sufficient quantities)
And about 300 other, less common, variants. (Some cause problems, some don't.)
ANEMIAS OVERSIMPLIFIED (see also Lancet 355: 1169 & 1260, 2000)
Anemia is any reduction below normal limits of the total circulating RED CELL MASS.
All anemias except the anemia of hyperacute blood loss (why?) are detected by your discovery of decreased hematocrit and/or decreased hemoglobin.
Hemodilution from excess fluids can occasionally lower hemoglobin and hematocrit in the absence of anemia. ("Pump him full of normal saline!!")
Acute anemia (i.e., blood loss) produces shock. (Hemoglobin and hematocrit stay normal or near-normal until plasma volume is restored.)
Chronic anemia requires increased cardiac output and eventually may produce hypoxia, weakness, malaise, easy fatiguability, koilonychia (supposedly). The fatty change in the myocardium ("tiger stripe"; "thrush breast") and liver is only seen when anemia gets really severe.
ANEMIAS OF BLOOD LOSS
Acute blood loss:
Remember that hematocrit and hemoglobin will remain normal until plasma volume is restored (hours to days.)
The bone marrow can increase erythropoiesis to three times normal within a day or so.
Chronic blood loss:
You'll see plenty of people with longstanding bleeding ulcers, GI and GU cancers, very heavy periods, and so forth on rotations. You'll learn about the special case of twin-twin transfusion in "Repro".
Anemia usually develops only when iron stores run out, i.e., iron deficiency anemia results.
Remember that hemorrhage is much commoner than hemolysis!
HEMOLYTIC ANEMIAS: INTRODUCTION
![]() Hemolytic anemia
Hemolytic anemia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
Many anemias are due, in whole or part, to premature destruction of red blood cells within blood vessels (INTRAVASCULAR HEMOLYSIS, as in DIC, or when RBC's are sensitized to complement or mechanically injured or lysed by oxidative stress in G6PD deficiency) or more commonly in the RE system (EXTRAVASCULAR HEMOLYSIS, as when RBC's are too stiff or fragile or are altered immunologically; in ongoing extravascular hemolysis, expect a biggish spleen). Easy update: Am. Fam. Phys. 69: 2599, 2004.
IgM sensitized red cells are usually destroyed intravascularly; IgG sensitized red cells are usually destroyed extravascularly.
Falciparum malaria often produces intravascular hemolysis; the other malarias usually produce extravascular hemolysis.
If the capacity of the ERYTHROID (red-cell producing) bone marrow to produce red cells is exceeded (it can increase output up to maybe eight times normal), anemia will result.
Regardless of cause, ongoing hemolysis will result in several pathophysiologic changes:
(the gut knows about the increased erythropoiesis and absorbs more iron -- diminished hepcidin --; if hemolysis is the only problem, this is seldom serious; look for iron in the Kupffer cells of the liver)
(this is a measure of the percentage of very young red blood cells; do this FIRST in your workup of anemia. Savvy labs now do it for you on any patient without a known cause of the anemia: Am. J. Clin. Path. 124: 129, 2005.

(in children, this produces thickened diploe of the skull, with a "crewcut" pattern)
If RBC's are being destroyed in the spleen (the usual site of extravascular hemolysis), it will usually be large, and is often iron-loaded and thus quite brown.
HEREDITARY SPHEROCYTOSIS
|
|
This is a relatively common autosomal dominant hemolytic syndrome (1/5000 people of northern European stock).
There are several loci, the most familiar being mutated spectrin.
* "Big Robbins" is confusing about compound heterozygosity in spherocytosis. Around 75% of cases are autosomal dominant, the rest mostly autosomal recessive at the ankryn, spectrin, or 4.2 loci and of course tend to be more severe, often obvious in newborns. They are mostly "compound heterozygotes" since there are a host of weak-abnormal alleles that seldom match (Semin. Hemat. 41: 118, 2004.)
Anemia is moderately severe, with hemoglobin around 9.
Poor binding of protein 4.1 to spectrin results in a defective erythrocyte cytoskeleton. This causes loss of the lipid bilayer membrane over the first few days in the bloodstream, resulting in spherical RBC's that get destroyed in spleen.
Spherocytes are easy to identify on a smear of peripheral blood because they lack the central pallor of biconcave RBC's.

In addition, the mean corpuscular hemoglobin (MCHC) is HIGH in this disorder because the spleen nibbles bits of membrane off the surfaces of RBC's that make it through.
The inadequate cytoskeleton allows the RBC's to rupture easily when placed in mildly hypotonic solution (increased osmotic fragility). All but the youngest RBC's are destroyed while passing through the splenic sinusoids.
Hereditary spherocytosis patients may have hemolytic and/or aplastic
(parvo B19)![]() crises, like sickle-cell disease
patients.
crises, like sickle-cell disease
patients.
|
|
Treatment is splenectomy, which results in a great subjective benefit and a normal hemoglobin (though some hemolysis continues.)
In HEREDITARY ELLIPTOCYTOSIS ("hereditary ovalocytosis"), the spectrin (usually) mutation is different (Blood 109: 3538, 2007) , the cells are oval, but the picture is the same or milder. It's actually more common than spherocytosis and is extremely common in Malaysia (their mutation, yes, gives resistance to malaria), but is usually not bad enough to cause anemia.
![]() Ovalocytosis
Ovalocytosis
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
* Variation on the theme: the rare "hereditary pyropoikilocytosis" is a hemolytic anemia with a mutant spectrin that won't stick to itself and gets degraded too rapidly. The cells have bizarre shapes ("like in a burn patient" -- hence the weird name), the anemia is obvious at birth, but the prognosis is good. The alpha-spectrin allele: Blood 106: 4367, 2005.
HEMOLYTIC DISEASE DUE TO ERYTHROCYTE ENZYME DEFECTS
Inherited disorders of enzymes in the hexose monophosphate shunt (glutathione synthetase, G6PD), other glutathione pathways, or glycolytic (hexokinase, pyruvate kinase) enzyme systems in erythrocytes result in oxidative damage to RBC's.
Deficiencies of each enzyme may be mild or severe, the thus the clinical pictures are widely variable.
RBC's cannot generate the reducing power (i.e., reduced glutathione) required to repair oxidative / free radical damage to their hemoglobin and membranes.
Masses of oxidized, denatured (i.e., scrambled S-S bridges from free radical damage) hemoglobin form in the cells and are visible by special staining procedures (* crystal violet) as "Heinz bodies".
The spleen futilely pits these out, making "bite cells." These are excessively fragile and get destroyed even more readily, making the anemia worse.
"Methemoglobin" carries at least one Fe+3 ion. This makes it brown and also increases affinity of iron to both the ferric ion and the other ferrous ions that may be left in the molecule.
GLUCOSE-6-PHOSPHATE DEHYDROGENASE deficiency (Am. Fam. Phys. 72: 1277, 2005) is the best known of these disorders.
This is X-linked and very common in certain populations. (Ten percent of US black men have it.)
In the commonest African variant (G6PD-), only the older RBC's are seriously deficient. In the commonest Mediterranean variant (G6PD Mediterranean), there is little good G6PD in any RBC.
Oxident stress (even the reactive oxygen species generated during infection) lyses red cells lacking G6PD. Famously, victims must avoid certain drugs (the basic "treatment", much better than trying to re-reduce the patients' hemoglobin).
This was a serious problem for black servicemen taking antimalarials in Vietnam. Sulfas, nitrofurantoin, and even aspirin can be hazardous for these people, depending on how badly deficient they are.
Some folks with G6PD get massive acute hemolysis from fava beans; some do not. We don't know why -- it doesn't seem to be particular alleles, and as affected children grow up they often lose the trait (Int. J. Heme. 83: 139, 2006). Part of the problem is that we can't test prospectively by passing out fava beans to test subjects and seeing whether they get life-threatening hemolysis. Stay tuned -- I believe there is a second metabolic defect.
Following exposure to the offending oxidizer, the older red cells are hemolyzed. The exposure can then usually continue without the patient remaining super-sick, as the vulnerable red cells are gone. Be sure you understand this. Since hemolysis in G6PD deficiency is intermittent, the big spleen and gallstones seen in other hemolytic diseases usually don't develop.
If the patient also has Gilbert's, severe neonatal jaundice results (Proc. Nat. Acad. Sci. 94: 12128, 1997). The combination can also occur later in childhood (Ped. Hem. Onc. 22: 561, 2005), or G6PD deficiency without Gilbert's can cause severe jaundice, with or without anemia, in babies.
Deficiency of PYRUVATE KINASE (for anaerobic glycolysis) is a very common Amish autosomal recessive birth defect. (Patients are mostly of northern European ancestry.) There are other mutations as well. Some molecular biology: Blood 82: 1652, 1993; Blood 89: 1793, 1997.
There is not enough energy to run the RBC membrane sodium pump, so the cells eventually undergo osmotic lysis.
Because these people have so much 2,3-DPG in their erythrocytes, they tolerate their anemia very well.
SICKLE CELL DISEASE (Ped. Clin. N.A. 49: 1193, 2002; Lancet 376: 2018, 2010)
![]() Sickle Cell Anemia
Sickle Cell Anemia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
Sickle cell substitution of valine for glutamine at the sixth position of the β chain (βS). Two α chains plus two βS chains makes HgbS.
Eight percent of blacks in the US are carriers, and 1 child in 600 is affected. There are around 100,000 sickle cell patients in the US.
In Britain and elsewhere, there has proved to be little demand for screening among pregnancies in populations at risk; people are now talking about being far more aggressive about reaching these people (BMJ 341: c5132, 2010).
Deoxygenation results in tactoid formation ("crystallization", "gelation") of HgbS. This forms sickle-shaped cells, and results in stasis (sludging), vaso-occlusive phenomena, and hemolysis.
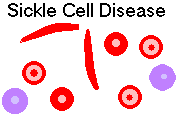
Sickling of a cell is reversible until the tactoids wreck the cell membrane ("how" involves spectrin tetramers dissociating and re-associating: Blood 81: 522, 1993). Then it's irreversible and the cell eventually gets lysed. Red cell life span averages about twenty days.
Dehydration increases MCHC and promotes sickling.
Hgb F effectively prevents sickling, so patients don't sickle until they are around two years old and no longer have much Hgb F. Some genotypes at other loci keep a lot of Hgb F around through life, and this makes SS disease far milder (Blood 118: 19, 2011).
Hgb A inhibits sickling, so heterozygotes (Hgb AS) do not have much trouble with sickling at usual oxygen tensions.
Hgb C does not inhibit sickling so well as Hgb A. Hgb SC mixed heterozygotes do sickle, though it is not so severe as Hgb SS homozygotes, and the spleen is typically lost later in childhood (Blood 85: 2238, 1995).
When oxygen tensions are low, sickled cells tend to stick tight to one another. This doesn't help things. Nobody really understands why this happens; some people think the sickled cells grow tiny spikes and stick together like burrs / velcro (described classically in Blood 81: 3138, 1993). Somehow the cells also get a lot stickier to endothelium, probably accounting for a lot of the strokes and other occlusive phenomena.
Decreased hemoglobin concentration helps prevent sickling. Concurrent iron deficiency anemia or thalassemia relieves sickling some, but staying very well hydrated probably helps even more.
Patients with sickle-cell anemia have many problems.
They have a severe, chronic hemolytic anemia with all the associated pathology.
Vaso-occlusive complications include leg ulcers, strokes (mechanisms: Blood 89: 4591, 1997; prevention and treatment Arch. Neuro. 58: 565, 2001), retinal infarctions, priapism (Am. J. Med. 94: 289, 1993), painful bone infarcts (necrosis of the femoral head -- J. Bone Joint Surg. 88: 2563, 2006, etc.), thrombi. A host of studies in the basic biology of veno-occlusive crises are available. We've known for some time that a likely contributing factor is double-stranded RNA, which glues sickled cells to endothelium, which probably explains why viral infection precipitates these events (Blood 85: 2945, 1995). Watch for endothelin-receptor antagonists (J. Clin. Inv. 118: 1924, 2008 -- keeps the little vessels from constricting around the plug) and intravenous immunoglobulin (Blood 111: 915, 2008 -- keeps the neutrophils away); both seem to help in the mouse model.
As the red cells lose oxygen / go to areas with lower pH, they tend to sickle. This isn't a problem in most areas. However, flow of blood through the spleen and marrow tends to be slow. Sludging of cells in the spleen first enlarges it (early childhood) then red-infarcts it by age 6 ("autosplenectomy"). The major problem then is vulnerability to pneumococcal and H. flu ("encapsulated organisms")infections. This fact also explains the marrow necrosis that underlies some of the crises in sickle cell disease.
Infections, notably salmonella arthritis / osteomyelitis (and even sepsis: J. Ped. 130: 349, 1997), are common; the bone and joint infections are vicious and often lead to osteonecrosis (Clin. Ortho. 468: 1676, 2010).
"Chest syndrome", with fever, chest pain, a lung infiltrate, and leukocytosis, is still not altogether understood ()NEJM 342: 1855, 2000; morphine as a cause Clin. Pharm. Ther. 75: 140, 2004). Occlusion of the vasculature is probably the key, especially embolization from infarcted marrow; it also often runs with a chest infection. It's pretty much impossible to tell it from pneumococcal pneumonia or pulmonary infarct, and it's treated as if it were pneumonia; it remain the leading cause of death in sicklers. Controlling costs (~$30,000 per admission) by a protocol: Pediatrics 127: e480, 2011.
![]() Sickle cell lung
Sickle cell lung
Big case; follow the arrows
Dr. Warnock's Collection
Hemolytic crises are triggered by exertion, infection, etc., while aplastic crises can result from severe
infection (typically due to parvovirus B19![]() -- review Blood 103: 422, 2004) or (maybe) folic acid deficiency.
-- review Blood 103: 422, 2004) or (maybe) folic acid deficiency.
* Future pathologists and radiologists: For some reason, the walls of the marrow arteries collagenize in sickle cell disease, getting progressively more striking as the person grows older (Arch. Path. Lab. Med. 128: 634, 2004).
* Survival is improving; the chance of a child with sickle cell disease dying is a bit less than 1% per year (Blood 103: 402, 2004); the rate of stroke is about the same.
These people often become addicted to opiates, and come to be despised by health-care providers as "drug seekers" or worse. Stigmatization of sicklers is unfair and scandalous (BMJ 318: 1585, 1999). They present the same kind of pain management problems as do cancer patients, only it is worse, since they live longer. Physicians are just now starting to get wise to this (Ped. Clin. N.A. 47: 699, 2000); managing it appropriately seems to reduce hospitalization by about half (Ann. Int. Med. 116: 364, 1992; epidural fentanyl is eminently sensible Pediatrics 93: 310, 1994).
About half of sicklers survive to age forty. Other genes modify the severity of the disease, and are the major influence on survival (NEJM 330: 1639, 1994.
There are reports that zinc deficiency (urinary loss? impaired GI absorption?) is a common, serious problem in these people, resulting in reversible drops in some lymphocyte populations (J. Clin. Lab. Invest. 130: 116, 1997). Some folks are now routinely supplementing sicklers with zinc (J. Lab. Clin. Med. 152: 67, 2008).
Sickle cell carriers (heterozygotes, "sickle cell trait", Hgb AS) trait have about 40% Hgb S. It is asymptomatic unless the patient goes to a high altitude (i.e., un-pressurized aircraft, too little oxygen in the anesthetic mixture, etc.), undergoes intensive physical training when out of shape or in the heat or dehydrated or overdressed (Am. Fam. Phys. 54: 237, 1997; update AJFMP 30: 204, 2009; potential causes of sudden death), or develops hematuria.
As you recall, heterozygotes are relatively protected against falciparum malaria, which is of course why hemoglobin S is so common in Africa (reconfirmed JAMA 297: 2220, 2007; mechanism is at least in part enhanced phagocytosis of ring-parasitized red cells with sickle or thal Blood 104: 3362, 2004). However, the protection is by no means absolute, and malaria remains a major killer of children with sickle-cell disease in Africa: Arch. Dis. Child. 89: 572, 2004.
"Big Robbins" could have mentioned that sickle cell trait tends to destroy the renal medulla by the end of the teenaged years. The military discovered during the Vietnam era that sickle carriers tended to die suddenly during the roughest parts of basic training or when they became stressed in the heat; today the military just makes an extra effort to keep these people well-hydrated which seems to solve the problems. Sports participation (especially college football) for these kids was the subject of a series of lawsuits in the past few decades; in April 2010, the NCAA (as part of the settlement of the Dale Lloyd case) agreed to mandate that all college athletes be screened for sickle cell and those testing positive be given informed consent and kept well-hydrated.
You will learn cost-effective screening procedures for sickle cell disease and trait later in your career.
Screening newborns for sickle cell disease allows early administration of pneumococcal vaccine, a potentially life-saving intervention. For black newborns, the extra cost is only $3100 more per life saved than not screening (J. Ped. 118: 546, 1991).
* We wisely screen all newborns. There's a fallacy in the above article ("$450 billion per life saved screening low-prevalence non-black populations" -- anybody can check the wrong box, and there's no shortage of Sicilians and Chinese ancestries.)
* Diagnosing sickle cell disease in the unborn at 10 weeks using PCR on Mom's blood rather than amniocentesis: Nature Genetics 14: 264, 1996.
Nonablative marrow transplant from HLA-matched siblings is now giving what seem to be excellent results, with patients free of both the disease and graft-versus-host: JAMA 312: 48, 2014. I'm in agreement with the NIH team that thinks both children and adults deserve this curative therapy (Blood 118: 1197, 2011.) Adults may benefit from non-myeloablative therapy -- they become chimeras for normal red cells.
More about treating the hemoglobinopathies:
Cord-blood allografts from HLA-identical newborn sibs followed by administration of growth factors have cured sickle cell disease (J. Ped. 128: 241, 1997). It is now routine when there is a matched sibling (Lancet 360: 629, 2002).
Allogenic stem-cells is curative in children; adults generally cannot tolerate it. Update NEJM 361: 2309, 2009.
Gene therapy is approaching -- for example, modified autologous stem cells cure mice (Blood 108: 1183, 2006.)
Hydroxyurea / hydroxycarbamide (Blood 79: 2555, 1992; Medicine 75: 300, 1996; Blood 115: 5300, 2010, a ribonucleotide reductase inhibitor) enhances the fetal hemoglobin response (nobody knows how) and diminishes the ability of red cells to sickle. Physicians have been reluctant to prescribe it long-term but the Greeks have found that it seems to improve length and quality of life into adulthood (Blood 115: 2354, 2010). The effect can be augmented nicely by erythropoietin (NEJM 328: 73, 1993).
* Watch for inhibitors of the gardos channel, which modules entry of water into the cell; these might well prevent sickling itself. The inhibitor is ICA-17043 (HemeOnc Clin. N.A. 19: 975, 2005). Study still underway.
* In the early 1990's, Illinois's 1183 sickle cell patients had a median of 3 hospital admissions yearly and consumed $59,000,000 in two years, or about $30,000 per patient per year; national numbers are similar (Pub. Health. Rep. 112: 38 & 44, 1997; they talk about strategies to improve out-of-hospital compliance, doctors' efficiency).
Sickle cell disease is undertreated. After reviewing JAMA 312: 1033, 2014 (standards for treating sickle cell) I was puzzled, as the authors seem to have been, about why most / all sicklers aren't on hydroxyurea and getting frequent transfusions.
HEMOGLOBIN C DISEASE
Hemoglobin C is another hemoglobin of African origin, almost as common as Hgb S. The carrier state and disease seem to protect particularly against cerebral malaria (JAMA 297: 2220, 2007).
People with Hgb CC semi-disease have some hemolysis and maybe a mild anemia, while people with Hgb SC disease have a sickle cell disease.
The diagnosis is often obvious from the peripheral smear.
Any patient with Hgb C, even a carrier, will have many target cells on peripheral smear.
Patients with Hgb CC disease have rod-shaped hemoglobin crystals in their red cells (see at low pH), while patients with Hgb SC disease have irregularly-shaped crystals ("birds in flight," etc.)

HEMOGLOBIN E DISEASE
A Southeast Asian hemoglobin (* glutamine for lysine at position 26 of the β chain) that produces mild hemolysis in homozygotes.
The mutation causes some underproduction of β chains, so if the other β gene is for a β-thalassemia, the patient can expect to have something on the β-thal spectrum.
THE THALASSEMIAS ("Cooley's anemia", "Mediterranean blood", etc.; Am. Fam. Phys. 80: 339, 2009; Lancet 379: 373, 2012)
Disorders with decreased synthesis of a structurally normal globin chain.
The other chain is made in normal quantities. Aggregates of this chain accumulate in the normoblasts and cause intramedullary hemolysis. (Beta chains are worse than α chains in this respect.)
I will resist the temptation to talk about the molecular genetics of the thalassemias except in the most basic terms. Those seeking an appreciation of the range of defects may begin with Mayo Clin. Proc. 76: 285, 2001; Clin. Chem. 46: 1284, 2000.
The thalassemias, like sickle cell disease, hemoglobin C, hemoglobin E, and G6PD deficiency, bestow resistance to falciparum malaria on their carriers (update Am. J. Hum. Genet. 77: 171, 2005).
ALPHA-THALASSEMIA: Asian and African genes. Usually the genes are deleted.
There are two genes for the α chain per chromosome, or four genes total. In the various α thalassemias, one or more are hurt.
One gene hurt ("silent carrier"): No health problem; red cells may be on the small side; maybe 3% Hgb Bart's (four γ chains) at birth, no Hgb H (four β chains) as adult.
Two genes hurt ("α thalassemia minor" or "trait"): Red blood cells tend to be small (MCV 82 or so) but anemia is unusual; 5-10% Hgb Bart's at birth, trace of Hgb H as adult. Seldom noticed.
We have classically screened for alpha thal trait by looking for Hgb H ("Bart's carriers"). A new method, which is under study, looks for the obscure "zeta globin", elevated in these folks (Am. J. Clin. Path. 129: 309, 2008).
Three genes hurt ("Hgb H disease"): Hemolytic anemia throughout life; 25% Hgb Bart's at birth; 25% Hgb H as adult. Hgb H is unstable and these patients have mostly a "bite cell" anemia, Heinz bodies and hemolysis. It also have excessive affinity for oxygen, and does not give it up easily enough for optimal tissue function, so the tissue hypoxia is worse than you'd expect for the degree of anemia. It's fairly well-tolerated, depending on exactly what mutations are present (NEJM 364: 710, 2011).
Four genes hurt ("hydrops fetalis"): Fetus dies in third trimester as it has only Hgb Bart's, which has an excessive oxygen affinity and also has lysis of normoblasts in the marrow due to four-gamma tetramers. (A novel hemoglobin with two zeta chains and two gamma chains in the early fetus allows survival for a while.) Death is due to congestive heart failure due to anemia and ineffective oxygen delivery to the tissues, so the fetus dies extremely edematous. (The other common causes of such a severe anemia are parvo 19 and Rh incompatibility.)
![]() Hydrops fetalis
Hydrops fetalis
Intrauterine death from Rh disease
KU Collection
* The crew at Brown, your lecturer's alma mater, brings a fetus with Bart's hydrops to term and are going to bone-marrow-transplant.... (Ob. Gyn. 85: 876, 1995). Your lecturer sees this as a prime example of the "law of inverse care"; you may disagree.
NOTE: It's desirable for a person with sickle cell disease or trait to have deletions of an α gene or two (why?) See J. Clin. Invest. 88: 1963, 1991.
BETA-THALASSEMIA: Mediterranean genes, mostly
There is one gene for the β chain per chromosome, or two genes total. In the various beta thalassemias, there is a problem with the mRNA and enough β chains do not get produced.
(Alleles: β0 -- no chains produced; β+ -- some chains produced, but not enough)
Heterozygotes ("beta thalassemia minor" or "trait"): mild or no anemia; hypochromia and microcytosis are usual, but target cells, teardrops and basophilic stippling are seen only in a minority of cases (Am. J. Clin. Path. 129: 466, 2008). You may not be able to appreciate either the hypochromia or the microcytosis on smear, because the cells tend to assume a pancake shape, without central pallor. Hgb F is sometimes increased; Hgb A2 is usually increased (because these have no beta chains, of course -- and this is the ONE time you might actually pick up "delta thalassemia", a lab curiosity with problems making delta chains, since your "beta thal trait" non-patient lacks the usual increase in Hgb A2.) It's VERY important to pick up your beta-thal-minor patients for genetic counselling and so that somebody else doesn't treat them for "iron deficiency anemia".

Homozygotes ("beta thalassemia major"): severe anemia beginning in infancy as the baby switches from Hgb F production. Patients have the stigmata of chronic hemolysis ("crewcut" skull x-ray, etc.), and little or no Hgb A, with extra Hgb F (usually most of the hemoglobin in Hgb F) and sometimes extra Hgb A2. As they must be transfused repeatedly and over-absorb iron due to ineffective erythropoiesis suppressing hepcidin, they have classically died of iron overload in their teens. The iron chelators (deferiprone, desferrioxamine, deferasirox -- the new one, Blood 116: 537, 2010 -- others) have been a great help, and now these people will probably live out a normal lifespan of good quality. The current major problems with the chelators (especially deferiprone) have been joint damage, agranulocytosis and zinc deficiency; (no surprise): Blood 80: 593, 1992; NEJM 332: 918 & 953, 1995; Br. Heart. J. 73: 486, 1995; Blood 90: 994, 1997. Marrow transplant: Arch. Dis. Child. 66: 517, 1991. Improved long-term results without bone marrow transplant: Blood 104: 34, 2004.

* "Beta thalassemia intermedia" can result from two not-so-bad β+ genes, or thal minor with extra copies of alpha chain. It is a fairly severe illness (genetic screening Arch. Dis. Child. 72: 408, 1995).
Don't worry about such arcana as "hereditary persistence of fetal hemoglobin", "δ-thal", etc., etc. Blood 80: 1582, 1992.
Every once in a while, one or more globin genes can be lost in the myelodysplastic syndrome, producing an acquired thalassemia (Blood 103: 1518, 2004).
* Mainland China's pathologists develop and begin implementing "preventive genetics" to eliminate the devastating thalassemias: J. Clin. Path. 57: 517, 2004. This isn't the place to talk about the "ethical" and "human rights" implications; the mere fact that we in the United States CAN do so is a real privilege.
* Heads-up: Ignore "fine basophilic stippling." It is usually an artifact caused by slow-drying of a smear.
You'll frequently need to distinguish the two great microcytic anemias: thalassemia and iron deficiency. In the US, they're about equally common; remember that there are a great variety of genetic thal-minor variants (Am. J. Clin. Path. 127: 192, 2007). A variety of formulas exist to give you the first guess. All are based on the fact that thalassemia cells tend to be smaller with a higher hemoglobin concentration, and iron-deficient cells tend to be larger with a lower hemoglobin concentration (Arch. Path. Lab. Med. 116: 84, 1992).
The formulas don't factor hemoglobin E (which one might consider a thal variant since the mutated beta-chain is sometimes undersynthesized) into the picture, since they were developed before our country was enriched by the Southeast Asian immigration. Easy to remember: Mild microcytic anemia with near-normal RBC count: Thal minor or hemoglobin E.
|
|
PAROXYSMAL NOCTURNAL HEMOGLOBINURIA ("PNH")
Abnormal sensitivity of RBC's to complement-mediated lysis, especially at low pH (i.e., while you're asleep).
The mechanism has been worked out, mostly, and is complicated. There is some sort of immune attack on the marrow, and cells evade the attack by mutating the gene (PIG-A) that makes an inositol-based anchor for a group of surface proteins, including CD59 that confers resistance to lysis by the body's own complement (NEJM 330: 249, 1994; J. Clin. Invest. 96: 201, 1995; Blood 85: 1640, 1995).
This disease also affects myeloid cells and megakaryocytes, proving it's a stem cell problem. (In addition, it sometimes turns into marrow failure, i.e., it's a Nowell's law hit. Or PNH may supervene when the immune attack on the marrow has already been underway as a known "aplastic anemia." How this happens: Ann. Int. Med. 136: 534, 2002.
Additional problems include the generalized toxic effect of a lot of hemoglobin floating free in the blood (soaks up nitric oxide that you need to keep the right vessels open), loss of iron into the urine (eventually producing iron deficiency) and thromboses especially in liver and brain veins (once attributed to abnormal platelets, it's more likely due to abnormal complement activation). Like all hemolytic processes, it can render people folic acid deficient.
The old "Ham Test" (acid hemolysis) tested the ability of complement to lyse red cells at low pH. Today's molecular diagnostics have replaced it (Am. J. Clin. Path. 114: 798, 2000). Today the diagnosis will probably be molecular.
Managing PNH is mostly about treating the symptoms, and hope it remits (i.e., your internal milieu changes somehow and there's selection against the bad clone, which is fairly common: NEJM 333: 1253, 1995). Treatment with the antibody eculizumab, which blocks the activation of C5 to C5a and the formation of membrane attack complex, is the breakthrough for PNH (NEJM 355: 1233, 2006; update Blood 111: 1840, 2008; Lancet 373: 759, 2009); this controls hemolysis and -- surprisingly, maybe -- thrombosis.
The alternative is bone marrow transplantation: Blood 113: 6522, 2009.
AUTOIMMUNE HEMOLYTIC ANEMIAS
WARM ANTIBODY AHA:
Extravascular hemolysis resulting from sensitization of RBC's to the patient's own IgG (sometimes IgA). Portions of the membrane are removed bit by bit ("partial phagocytosis"), eventually imparting the spherocyte shape.
A specific cause is found in about one third of cases.
Check for cancers (especially lymphoma-leukemia), systemic lupus (lupus autoimmune hemolysis Am. J. Med. 108: 198, 2000), drugs (back when methyldopa was the most-prescribed medication in the US, 10% of patients receiving it became Coombs-positive and 1% had serious hemolysis.)
Evans syndrome is an often-severe combination of autoimmune hemolytic anemia and either isoimmune thrombocytopenia (ITP) and/or immune neutropenia (Blood 114: 3167, 2009.)
Mononuclear phagocytes and splenic macrophages first nibble at the sensitized membrane, turning the RBC's into SPHEROCYTES. Eventually the cells are destroyed.
DRUG HEMOLYSIS mechanisms in warm autoantibody disease:
HAPTEN MECHANISM (high-dose penicillin type): the patient makes an antibody against drug-red cell membrane complex
IMMUNE COMPLEX MECHANISM (quinine-quinidine type): drug-antibody complexes get absorbed to innocent bystander RBC's, which are then lysed by complement. Phenacetin and cephalosporin are also frequently implicated in this type of hemolysis.
AUTOANTIBODY MECHANISM ("Aldomet" type): the drug somehow causes one to make antibodies against one's own Rh antigens (everybody has some Rh antigens, even though Rh negative).
COLD AGGLUTININ AHA (Blood 122: 1114, 2013)
Due to IgM antibodies, which work best below 30.
These monoclonal auto-antibodies appear mysteriously in mycoplasma pneumonia (* anti-I), infectious mono (* anti-i), less often HIV, influenza, others. The anemia, if any, is self-limited and seldom detected.
Chronic cold agglutinin disease is fairly common in lymphoma, or may be idiopathic. (These people should dress warmly.) Curiously, hemolysis occurs in the liver, not the spleen, in these patients.
* The presence of cold agglutinins is a major nuisance for blood bankers trying to type blood. Let us worry about it.
COLD HEMOLYSIN AUTOIMMUNE HEMOLYTIC ANEMIA
The usual disease is PAROXYSMAL COLD HEMOGLOBINURIA and the auto-antibody is * Donath-Landsteiner auto-antibody, a polyclonal IgG against P blood-group antigen on the RBC's.
The classic story is the patient goes skiing and falls in the snow; the next time, after rewarming (complement works when it's warmer), the patient voids, the urine is dark brown.
Once a famous sign of syphilis![]() ,
paroxysmal cold hemoglobinuria now it most often follows the flu or
arises mysteriously. It's actually quite common in children now that we've started
looking for it (in fact, it's the most common autoimmune hemolytic anemia), and it's
occasionally a genuine side-effect of immunization.
,
paroxysmal cold hemoglobinuria now it most often follows the flu or
arises mysteriously. It's actually quite common in children now that we've started
looking for it (in fact, it's the most common autoimmune hemolytic anemia), and it's
occasionally a genuine side-effect of immunization.
Surprisingly, in today's era of body cooling during cardiopulmonary bypass, the disease seldom turns up. One case: Ann. Thoracic Surg. 75: 579, 2003.
HEMOLYTIC ANEMIA RESULTING FROM TRAUMA TO RED CELLS
![]() DIC
DIC
Schistocytes, no platelets
Wikimedia Commons
This can result from:
Fragmented RBC's on the peripheral smear (helmet cells, triangle cells, target cells, schistocytes)
These hemolytic anemias seldom last long, but indicate serious disease.
* "March hemoglobinuria" is another cause of intravascular hemolysis from trauma; it is well tolerated. "Burr cells", with little projections, are supposedly seen in patients with uremia (i.e., symptomatic kidney failure.)
![]() Burr Cells in Uremia
Burr Cells in Uremia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
MEGALOBLASTIC ANEMIAS
This is a broad term for all problems in which the normoblasts (and, often, the other cells in the body) cannot synthesize DNA fast enough to keep up with the growth of their cytoplasm. ("Nuclear-cytoplasmic asynchrony". This is easily the most plausible explanation; remember you also need B12 to do methionine and methyl-malonic acid). Although patients will be anemic, the baby cells (-"blast") end up big ("megalo-"), and a good pathologist can immediately recognize a "megaloblastic marrow smear" by the big, blue cells with lots of cytoplasm and immature-looking nuclei. (Polys and their precursors are big, too. You'll see pictures; I won't ask you to make the distinction on an exam.)
Regardless of the cause, expect to see:
Rule: Count one hundred neutrophils. Separate segments are defined to be masses separated by a thread composed entirely of heterochromatin. If you see one neutrophil with six segments, or five with five segments, you have established the diagnosis of megaloblastic anemia. I am suspicious when I see a three-lobed eosinophil does it, too.

PERNICIOUS ANEMIA ("true pernicious anemia", "addisonian pernicious anemia", etc.; Medicine 85: 129, 2006)
This is extremely common, especially in older folks; one group argues that there must be 800,000 undiagnosed cases (Arch. Int. Med. 156: 1097, 1996). Once dreaded and very lethal ("pernicious"), the available of injectable B12 (in the old days, a pound of raw liver per day by mouth, or liver injections) has now rendered this common problem almost innocuous.
The basic problems is chronic atrophic gastritis with destruction of the parietal cells and failure of intrinsic factor production.
Often (maybe 40% of the time) there is some antibody that sticks to intrinsic factor as well, preventing its binding to B12 ("blocking antibody"). There's also likely to be antibodies against receptors in the ileum where the complex must be absorbed ("binding antibody"). Assay J. Clin. Path. 46: 45, 1993 (* contrary to older sources, either can be present without the other).
* The antibodies in "pernicious anemia":
* While we are ordering unnecessary lab tests, most of these patients have elevated serum gastrin levels, too (why?). Despite the fact that we can test the antibodies, the main problem seems to be T-cells that destroy the gastric mucosa, with the antibodies being an epiphenomenon.
On rotations, be aware that it now seems that in pernicious anemia, the serum B12 level may be normal. Check the serum methylmalonic acid and homocysteine levels if you have reason to suspect B12 and/or folic acid deficiency; they are likely to be up and some say this is a good screen.
In addition to the problem making good blood cells, B12 deficiency (from whatever cause) is noxious to the brain and cord, probably because of increased levels of methyl-malonic acid and propionic acid. Demyelinization of the posterior columns of the spinal cord happens early and causes loss of proprioception and some paresthesias, as in tabes dorsalis. Eventually, dementia occurs. These problems may precede the anemia (Postgrad. Med. 91: 231, 1992; Postgrad. Med. 88: 147, 1990).
In the 1980's, I had the rare privilege of autopsying a "virgin" pernicious anemia patient, a gentleman who was institutionalized for five years with "Alzheimer's disease". His physicians drew blood from time to time, but never noticed the anemia, the MCV of 140, or the hypersegmented neutrophils reported by the lab. Bad care. I noted each of the following "classic" changes:
NOTE: Often the lateral columns are affected, too, though less dramatically. This is called "subacute combined degeneration of the cord", typical of B12 deficiency.
To make the diagnosis, we used to perform the two-part SCHILLING TEST.
Give the patient an injection of normal B12 first (why?)
Then administer radioactive B12 by mouth. A healthy person will excrete the radioactivity in the urine. If your patient, for any reason, cannot absorb B12 via the gut, the urine will not be radioactive.
If your patient cannot absorb B12 via the gut, then give him or her a dose of radioactive B12 with intrinsic factor. If this causes the radioactivity to appear in the urine, you know the patient lacks intrinsic factor. If the radioactivity still fails to appear, you know there's some problem with absorption (i.e., one of those antibodies and/or some disease of the gut).
Okay. Alas, the long-time supplier of the isotope withdrew it from the market in 2001. Stay tuned. Nowadays, you'll probably confirm your clinical impression of addisonian pernicious anemia by ordering a serum anti-parietal cell antibody; maybe 70% of them are positive.
It's common knowledge that patients with autoimmune gastritis, whether or not they have pernicious anemia, are at substantial risk for gastric cancer (carcinoma, carcinoid; old papers Cancer 71: 745, 1993 and Arch. Path. Lab. Med. 113: 399, 1989). It's ironic that, while the Japanese endoscope almost everybody (and often finding the highly-curable early gastric cancer lesions), we're just now getting used to the idea of endoscoping pernicious anemia patients regularly. For some better-late-than-never common-sense see Gut 34: 28, 1993; endoscopy every five years, more often if dysplasia is found, seems reasonable (Gut 31: 1105, 1990; policy-makers take notice). There's been no resolution, and this seems weird in a time when "everybody" after a certain gets colonoscopies.
Classic "addisonian" pernicious anemia is primarily a disease of people of northern European ancestry, though any race can be affected. The sex ratio is about equal, which is unusual for an autoimmune disease.
"Juvenile pernicious anemia" mimics the adult disease in its hematologic and nervous system manifestations. These patients don't usually have autoimmunity, but instead are born without good intrinsic factor, or without receptors for the complex. Tip: Check the urine for methyl-malonic acid (why?)
OTHER CAUSES OF B12 DEFICIENCY
"Inadequate diet" today means vegetarianism, especially if extreme. "Vegans", who will take no food of animal origin, all get B12 deficiency in a few months unless they supplement. B12 deficiency is rampant among today's "amateur" vegans, many of whom are teens (Am. J. Clin. Nutr. 76: 100, 2002).
Today, many people report feeling better taking only limited amounts of animal products. This probably includes the 7% of entering Year I medical students who self-identify as vegetarians (J. Am. Diet. Assoc. 107: 72, 2007) -- many are religious/cultural; these people are seldom militants and not surprisingly, maybe half have given up vegetarianism by the time of graduation.
By contrast, cult-vegetarians, the ones most likely to get into trouble, are hard to deal with. ("I can't take that particular medication because I read that the gel-capsule is made from animals.") The one's I've known grandstand ("Meat is murder!"), choose to believe even the most obvious pseudoscience, and are hostile to real evidence-based medicine. The greatest danger is to their children, and this is now a major public health problem. More about this under Nutritional Disease.
If your whole stomach is gone after some big operation, you'll need B12 supplementation, of course. But this isn't true pernicious anemia.
Worth knowing -- if the salivary glands are lost, B12 deficiency results due to lack of salivary-produced * haptocorrin, which protects B12 in the stomach. If pancreatic insufficiency is present, proteases may not be sufficient to split the * haptocorrin from the B12 to allow it to complex with intrinsic factor.
* Trivia: The receptor that absorbs intrinsic factor in the ileum is called "cubilin".
If you have malabsorption (sprue, Whipple's, lymphoma, scleroderma, others), or if you have the fish tapeworm on board, or if you have a blind loop (post-surgery, duodenal diverticula) full of bacteria, or if your ileum is messed up badly by Crohn's disease, you may need B12.
There's a good serum assay for B12. It's expensive, but perhaps an occasional screen is worthwhile, more for the mental problems that the deficiency causes especially in the elderly. I'd prefer you start your anemia workup with a reticulocyte count first.
* Hey Doc! Want to keep your poorly-educated patients happy and coming back ($$)? Some physicians diagnose "pernicious anemia" wantonly, and prescribe monthly B12 shots, which are red (ooh, pretty), painless, inexpensive, and harmless. Further, once you start doing this, it'll be hard to prove the patient didn't originally need the treatment and doesn't need to continue. However happy this common practice may make people, I have a ninth commandment problem with it. I hope YOU do, too.
![]() Pernicious Anemia
Pernicious Anemia
Text and photomicrographs. Nice.
Human Pathology Digital Image Gallery
FOLIC ACID DEFICIENCY (ever know someone who called it "vitamin P"? "folic" means "from (green) leaves")
This is disturbingly common in the U.S., and not just in the alcoholics, oldsters, and babies cited in "Big Robbins". Contrary to your text, the deficiency need not be "gross". You need some vegetables in your diet every once in a while, and many Americans don't like them. (In the poor nations, many people can't afford them.) If you lack folic acid, you can't shuttle your one-carbon units (i.e., methyl and formyl groups) around.
In pregnancy, the fetus leeches out Mom's folic acid, which may already be in short supply. Likewise, in hemolytic disease and in carcinomatosis, folic acid supplies often drop, a "relative folic acid deficiency" is unmasked, and a superimposed megaloblastic state develops.
Phenytoin and the birth control pill are infamous for interfering with the absorption of folic acid. Of course, so can other malabsorption problems (sprue and even GI lymphoma can present this way). Hemodialysis takes the vitamin out of the body, too.
A blood assay for folic acid is available. Again, it's expensive. I'd prefer you not order these for all your microcytic anemia patients at the onset of your workup. Tip: You can also detect folate deficiency by assaying the urine for formimino-glutamic acid (FIGlu), a breakdown product of histidine that requires folate for further processing.
BEWARE. Supplementing a patient with pernicious anemia with big doses of folic acid will improve the hematologic picture (nobody knows why) and exacerbate the brain disease (nobody knows why). This is the main reason that only small amounts of folic acid are put in over-the-counter not-for-pregnancy vitamins.
B-12 AND FOLATE UNRESPONSIVE MEGALOBLASTIC ANEMIA
Obviously, anti-DNA chemotherapy will produce megaloblastic changes.
Occasional megaloblastic anemias respond well to vitamin B1 (thiamine) or vitamin B6 (pyridoxine). These are usually acquired, and probably reflect Nowell's law hits (i.e., there is an increased leukemia risk.)
Note: Heavy alcohol drinking, and sometimes liver disease (maybe) and hypothyroidism (maybe), will raise the MCV to maybe 110 fL. Strangely (especially with alcohol), these red cells tend to be round, while the megaloblastic anemias feature big oval red cells ("macro-ovalocytes"). Nobody knows why.
IRON DEFICIENCY ANEMIAS (NEJM 372: 1832, 2015)
|
|
The "average diet" contains 15-20 mg of iron daily, mostly as heme in animal products which is readily absorbed. This is plenty for a majority of people. However, many folks (growing children, women with heavy monthly blood loss or pregnancy, those losing blood from disease) need more than this, and plenty of people are on iron-poor diets ("I'm a vegetarian and I don't believe in pills!", poverty, and/or ignorance.)
You already know that iron is absorbed (in limited, regulated quantities) in the duodenum, shuttled around on transferrin, used in hemoglobin, myoglobin, and cytochromes, and the storage supply found to ferritin as the invisible short-term form ferritin and the copper-penny, Prussian-blue-stainable long-term form hemosiderin.
If you look in the marrow, you'll find zero stainable iron, and probably more than the usual number of red blood cell precursors though they're quite small. Rumor has it that they are more numerous because they take longer to develop and perhaps divide a few more times, accounting for the hyperplasia.
A person may become iron-deficient by:
| Heme iron is much better absorbed than iron from beans and so forth. Especially, there is disappointingly little usable iron in spinach; what's there is oxalate-bound and unavailable. |

|
Poor diet is seldom the sole cause in U.S. adults consuming an adequate number of greaseburgers weekly. However U.S. kids can and do become iron deficient, despite "Big Robbins". There is very little iron in breast milk, and children do become deficiency as a result. No one knows the "best" amounts and timing for supplementation (Ped. Clin. N.A. 48: 401, 2001). Perhaps one US baby in 11 is iron-deficient enough to be anemic from it (J. Ped. 154: 44, 2009).
Iron deficiency is one of the great hazards of pregnant women. Improving the iron status by oral supplementation goes a LONG way to improving neonatal health and even avoiding low-birth-weight (BMJ 346: f3443, 2014).
Despite a great deal of disinformation by the vegetarian community leadership, who cherry-pick obscure journals, it is a plain fact that it is difficult for vegetarians to get adequate absorbable iron in their diet. See, for example, Am. J. Clin. Nutr. 89: 1685-S, 2009 (soybeans / tofu are low in iron, but iron in pills made from soybean ferritin is as bioavailable as ferrous sulfate and vegetarians might accept it more readily.) Europe's adult vegetarians tend to be both iron and B-12 deficient, with corresponding abnormal labs (Eur. J. Haem. 69: 275, 2002). England's young vegetarians are much more likely to be iron-deficient than their omnivore peers (Pub. Health. Nutr. 6: 485, 2003). Even the midwives are delivering serious warnings to pregnant vegetarian women about multiple deficiencies, including iron (J. Midwifery Women's Health 53: 37, 2008). Competitive athletes, who are reality-oriented, know they need to take supplemental iron if they're vegetarians (Nutrition 20: 696, 2008).
Anemia of vegetarianism is likely to have normal indices, since patients are both iron deficiency and B-12 / folate deficient. An American vegetarian mother who does exclusive breast-feeding for the first sixteen months almost kills her child: Ped. Heme. Onc. 29: 74, 2007.
Not everyone can absorb oral iron well. People with celiac sprue, atrophic gastritis, and even bad helicobacter infections may not absorb oral iron well enough for it to be an effective treatment.
Doc: If you find iron deficiency, you MUST find the cause. It is cancer of the GI or GU system until proven otherwise.
Around 10% each of toddlers, teenaged girls, and moms are iron deficient, and of these, around half are anemic (JAMA 277: 973, 1997). Newer numbers: Almost 20% of kids are iron-deficient, with 5% anemic (Clin. Ped. 44: 333, 2005); easy to diagnose, but these kids are still not being followed up. Of course this is deplorable. An attempt by the English to alter the junk-food habits of their underclass through "education" was an expensive, complete failure (Arch. Dis. Child. 76: 144, 1997). Older folks with congestive heart failure and mild iron deficiency (even without anemia) do MUCH better when you pick it up and address it (Heart 100: 1414, 2014.)
Regardless of the cause, the iron-deficient patient eventually develops a hypochromic, microcytic (why?) anemia, which can be very severe.
You'll be impressed the first time you see tiny red cells with broad central pallor (more than the usual 1/3).
For some reason, elongated "pencil RBC's" are common in iron deficiency (no one knows why, but they are quite distinctive for true iron deficiency: Am. J. Clin. Path. 129: 466, 2008), and the platelet count tends to rise (also mysterious).
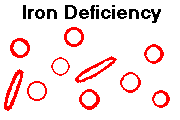
Textbooks describe a variety of additional physical findings that are supposed to be more or less specific for iron deficiency. This includes koilonychia (spoon-shaped nails), beefy "atrophic" glossitis (supposedly), intestinal malabsorption, and upper esophageal webs ("Plummer-Vinson"; the link to iron deficiency is almost certainly a myth, and I have my doubts about the others, since they make no sense physiologically).
Plasma ferritin levels usually closely correlate with total body iron, provided that the liver isn't acutely damaged. More about iron testing when we cover lab testing. In true total-body iron deficiency, serum iron is generally low (unless the patient just popped an iron pill for some reason) and iron binding capacity (basically transferrin) is high; transferrin saturation is way-low. Iron labs J. Cln. Path. 64: 287, 2011).
* Tip: Allied health professionals often refer to both hemoglobin and hematocrit as "serum iron". I've given up trying to explain.
ANEMIA OF (CHRONIC) INFLAMMATION ("Anemia of Chronic Disease", NEJM 352: 1011, 2005)
Here, there's plenty of iron in the body, but it isn't available to the normoblasts, but stays in the big fixed macrophage in the center of the erythroid island.
This unfortunate effect is mediated by increased body levels of interleukin 1 (* and probably other cytokines too), i.e., the macrophages are phagocytosing somewhere in the body, and the "acute phase reaction" has been going on long enough to cause lowering of the hematocrit.
Patients have a hypochromic-microcytic (if severe enough -- why?) anemia with a hemoglobin of around 8-10 gm/dL. Bone marrow iron stores are increased.
In the U.S., the usual causes are rheumatoid arthritis, osteomyelitis, TB![]() , and huge bedsores.
(Remember these also cause amyloidosis A; same underlying problem.) Treat the underlying cause,
Doc.
, and huge bedsores.
(Remember these also cause amyloidosis A; same underlying problem.) Treat the underlying cause,
Doc.
* If you want to diagnose and monitor the disease, try serial zinc protoporphyrins (Blood 81: 1200, 1993; why does it work?)
Artificial erythropoietin came into use for anemia of chronic disease during the late 1990's, and is now common.
SIDEROBLASTIC ANEMIA
Once again, there's plenty of iron in the body, and this time, it's even in the normoblasts. But in this relatively uncommon problem, patients have difficulty placing the iron into their heme rings. Instead, it remains in the mitochondria, which light up as Prussian-blue positive chunks in their ordinary position around the normoblast nucleus (hence the cute term "ringed sideroblasts").
Commonly, only some of the red cell precursors are affected (i.e., Nowell's law has been operating; this is a fairly common finding in the early myelodysplastic syndromes: update Blood 106: 247, 2005.) Alcoholism and isoniazid therapy also get cited.
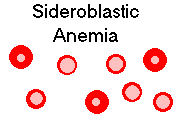
* In the X-linked hereditary sideroblastic anemia, the problem is with δ-amino levulinic acid synthetase (NEJM 330 675, 1994; Blood 90: 872, 1997; Blood 93: 1757, 1999). The gene is Abcb7: Blood 109: 3567, 2007.
Some other cases of sideroblastic anemia respond to big doses of pyridoxine, and in these cases, the pyridoxine binding site on the protein is what was hit by the mutation (of course). More on this: Blood 93: 1757, 1999.
PURE RED CELL APLASIA
BLACKFAN-DIAMOND SYNDROME (Blood 116: 3715, 2010; J. Clin. Inv. 122: 2346 & 2439, 2012) is the most familiar of the diseases with selective inability to make red cells.
Anemia may be macrocytic or normocytic and varies in severity.
Many of these patients have a mutation in the ribosomal proteins. The first discovered was abnormal ribosomal protein S19: Nat. Genet. 21: 169, 1999. There are at least 13 other loci, with heterozygotes (i.e., new mutations) affected (Blood 109: 1275 & 3152, 2007; Blood 112: 1582, 2008; Blood 117: 2567, 2011) -- the common theme is a problem with the translational machinery of the ribosome ("anemia lost in translation").
* It often responds to glucocorticoids and often remits with time. It remains quite mysterious. Marrow transplantation is curative.
THE FANCONI ANEMIAS, now numbering seventeen loci (2015) are recessive, thankfully rare illnesses. They feature apoptosis of erythrocyte precursors, as well as a predisposition to cancers (notably the myelodysplastic syndromes and acute myelogenous leukemia (there's a signature gain of parts of chromosome 1), but also head-and-neck squamous cancers and some other solid tumors). Often, there's total marrow failure in the end. Patients tend to be short and have other malformations, especially involving the thumbs. The products of most (Blood 120: 86, 2012) of these loci form an important protein complex ("the core complex") found in both cytoplasm and nucleus, and that is involved in repair of DNA crosslinks and the destruction of damaged cells. Update J. Clin. Inv.122: 3799, 2012.
Not surprisingly, mutations of the Fanconi loci are popping up in sporadic human cancers as well (Arch. Otol. 132: 958, 2006). Your lecturer predicts that mutations here will be markers for responses to particular chemotherapeutic agents.
CONGENITAL DYSERYTHROPOIETIC ANEMIA is a group of hereditary mostly-macrocytic anemias ("the CDA's") with delayed maturation of normoblasts (other lineages fine), which tend to be odd-looking and multinucleated. Several loci with typical syndromes are known (Blood 107: 334, 2006; Ann. Heme. 87: 751, 2008; Blood 117: 6928, 2011; Blood 122: 2162, 2013).
![]() Dyserythropoietic anemia
Dyserythropoietic anemia
Access Medicine
* Hypoplastic anemia and an extra joint in the thumb: Aase syndrome! Still being worked out.
* Another congenital anemia (autosomal dominant dyskeratosis congenita) lacks the ability to restore telomeres to the endlessly-dividing marrow stem cells (Lancet 359: 2168, 2002; Blood 102: 916, 2003; update J. Clin. Lab. Med. 162: 353, 2013).
In the chronic hemolytic disorders (notoriously sickle-cell disease, less often spherocytosis or hemoglobin C disease), the production of normoblasts can simply shut down, even when there's enough folic acid around. Parvovirus 19 is now known to be the usual cause -- it prevents normoblasts from maturing. This is called APLASTIC CRISIS.
| Future pathologists wishing to spot "parvo":
You will see no normoblasts beyond the basophilic stage,
and the basophilic normoblasts have the nuclear chromatin
pushed to the edge by the viral inclusion |

|
Patients with THYMOMA / THYMIC HYPERPLASIA often (7% for thymomas)develop a red-cell aplasia, probably because T-cells attack the normoblasts in the bone marrow. Removing the thymoma often solves the problem (Am. J. Clin. Path. 103: 346, 1995).
Patients who take CHLORAMPHENICOL can all expect some temporary suppression of erythropoiesis, and in 1 case in about 25,000 this is severe and permanent.
In KIDNEY DISEASE, there's often a lack of erythropoietin, and patients receive injections of the stuff, which helps.
FINISHING UP: Thankfully, hemoglobins with excessively high oxygen affinity ("Chesapeake" and many others) are uncommon. As you'd expect, these render the patient polycythemic.
* Red-cell substitutes have been a disappointment. A bovine hemoglobin preparation delivers the goods but only lasts for a day in the bloodstream; it is now in widespread use in some nations and helps JW's in the USA. The older perfluorocarbon emulsions ("Fluosol", others) required patients to breathe 100% oxygen which was deadly to the lungs. Review Am. J. Clin. Path. 118 S: S-71, 2002. Update from the Hop: Arch. Path. Lab. Med. 131: 734, 2007. Nothing else lately.
We'll do polycythemia vera, other secondary polycythemias, acquired (autoimmune / telomere troubles) aplastic anemia, and the myelodysplastic syndrome under "White Cells". Enjoy these pictures:
{13856} red cell, normal blood. (If platelets stick to polys, it's EDTA anticoagulant effect ("satellitism"). Some folks have a gene to make this happen.)
JEHOVAH'S WITNESSES AND BLOOD TRANSFUSION
Ask a spokesperson for the details. "Many Jehovah's Witnesses
feel that accepting a blood transfusion will lead them to eternal
damnation" (Ob. Gyn. 102: 173, 2003).
Here are some references that
will help you in your thinking.
On the one hand...
On the other hand...
The researchers were actually examining the old-fashioned, unscientific practice of
early blood transfusions for GI bleeders. Since the figure of 2.5% is astonishingly
low and comes from a paper from 1924, it may reasonably be questioned, and the authors
also note that the 1924 population was a selected group of young, physically-fit individuals
who may have gone straight to surgery.
The underlying causes of the bleeding aren't stated, but we may think that
the 1986 study included many inoperable cirrhotics, who typically bleed to death sooner or later
regardless of what we have to offer.
The JW's are apparently trying to trick the reader into believing
that the 1986 study was a comparison between GI bleeders who were
transfused and those who were not transfused. The researchers actually compared patients who were
transfused when they came in versus patients who were transfused only if their
hemoglobin dropped below 8 g/dL. The authors explain:
No scientific physician will be surprised that patients who were transfused
for no real reason ended up doing worse and requiring more blood in the long
run than patients for whom transfusion was delayed. You're just supplying
more pressure to keep the pumper open.
The authors finish up:
Go figure -- they must have also been sicker to begin with. The authors conclude from their study,
I think a reasonable person would agree that the purpose and findings of the
study should not be presented as part of an overall indictment of blood transfusion.
A group in Scotland extrapolated a 65x risk of fatal postpartum hemorrhage
(Arch. Gyn. Ob. 276: 339, 2007).
Sooner or later, you will run into the issue of blood for Jehovah's Witnesses.
The group is authoritarian, tightly-controlled,
and forbids blood transfusions for its members.
The British Journal of Surgery (October 1986) reported that prior to the advent of
transfusions, gastroinestinal hemorrhage had "a mortality rate of only 2.5 per cent." Since
transfusions became customary, "most large studies report a 10-percent mortality."
Why a death rate four times as high? The researchers suggested: "Early blood transfusion
appears to reverse the hypercoagulable response to haemorrhage thereby encouraging
rebleeding."
Of the 26 patients randomized to receive no blood transfusion within 24
hr of admission, five did require transfusion on their first day for anaemia
worse than 8 g/dl.
The role of blood transfusion in the treatment of gastrointestinal haemorrhage
has never been questioned seriously although it is accepted that
patients who are transfused more than 4 units of blood fare worse than those
who require less than 4 units.
Therefore, where possible, blood transfusion should be delayed.
If it does need to be given for severe haemorrhage, early surgery
should be considered.
Women who are Jehovah's witnesses are at a six times increased risk for maternal
death, at a 130 times increased risk for maternal death because of major
obstetric haemorrhage and a 3.1 times increased risk for serious
maternal morbidity because of obstetric haemorrhage, compared
to the general Dutch population.
{13858} red cell, normal marrow, unusual stain
{13976} red cell, normal blood (two orthochromatic normoblasts)
{20780} red cell, normal blood
{46538} red cell, normal blood
{26161} basophilic normoblast (prorubricyte)
{26162} basophilic normoblast (prorubricyte)
{26197} basophilic normoblast (prorubricyte)
{26198} basophilic normoblast (prorubricyte)
{26223} polychromatophilic normoblast (rubricyte)
{26224} polychromatophilic normoblast (rubricyte)
{18690} orthochromatophilic normoblast (metarubricyte)
{10127}red cell, even staining for hemoglobin
![]()
{10130} Heinz bodies
{10418} nucleated red cells in hemolytic disease of the newborn
{21113} hemolytic disease of the newborn
{13898} polychromasia (the purple, large red cells are "reticulocytes")
{14722} reticulocytes, normal
{14723} reticulocytes, too many
{39616} erythroid hyperplasia
{39617} erythroid hyperplasia
{27434} plasmodium vivax
{13880} cold agglutinins in the blood
{12299} hereditary spherocytosis
{16172} spherocytes
{11547} hereditary elliptocytosis (spherocytosis variant)
{13892} hereditary elliptocytosis (spherocytosis variant)
{12305} spur cells ("acanthocytes"' E.T.-finger cells); abetalipoproteinemia case;
there are other odd genetic syndromes featuring this anomaly
{13871} abetalipoproteinemia smear (lymphocyte in upper right)
{20128} abetalipoproteinemia smear
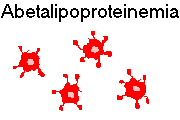
{17419} sickle cell disease
{13907} target cells
{16167} target cells
{16177} target cells
{16169} stomatocytes (flattened uniconcave RBC's typical of acute drunkenness, liver disease and
various rarities, everyone's allowed a few)
{12308} schistocytes
{13895} schistocyte
{16178} schistocytes
{17137} DIC
{20149} thalassemia
{13877} basophilic stippling
{10430} "megaloblasts" (i.e., the red cell precursors in pernicious anemia)
{13751} megaloblastic blood smear
{13754} megaloblastic marrow
{13757} megaloblastic marrow
{13889} macro-ovalocytes (i.e., megaloblastic anemia)
{12302} teardrops

{14013} sideroblastic blood (dimorphic RBC population)
{27342} ringed sideroblasts, iron stain (not the greatest photo)
{09054} iron in mitochondria
{14025} Pappenheimer body (retained iron-loaded mitochondrion, i.e., you have sideroblastic
anemia and/or have had a splenectomy; you need a Prussian blue stain to see them well)
{16168} Pappenheimer bodies
{13854} polycythemia vera blood
{13976} rouleaux
{14028} multinucleated red cell (DiGuglielmo's?)
{23920} myelodysplastic syndrome
{13883} Howell-Jolly bodies (stray chromosomes not extruded; usually mean the patient has had a
splenectomy -- say, after trauma, for treatment of bad thalassemia or spherocytosis,
or a sickler's autosplenectomy -- and they were not removed)
{16170} Howell-Jolly bodies
{13904} post-splenectomy
{29011} blood bank
![]() Howell-Jolly bodies and nucleated red
Howell-Jolly bodies and nucleated red
WebPath Photo
Let the young sing songs of death. They are stupid. The finest thing under the sun and the moon is the human soul. I marvel at the small miracles of kindness that pass between humans. I marvel at the growth of conscience, at the persistence of reason in the face of all superstition and despair. I marvel at human endurance.
-- Anne Rice, "Pandora" ("The Vampire Chronicles")
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about diseases of red blood cells to consult these standard textbooks.
In my notes, the most helpful current journal references are embedded in the text. Students using these during lecture strongly prefer this. And because the site is constantly being updated, numbered endnotes would be unmanageable. What's available online, and for whom, is always changing. Most public libraries will be happy to help you get an article that you need. Good luck on your own searches, and again, if there is any way in which I can help you, please contact me at scalpel_blade@yahoo.com. No texting or chat messages, please. Ordinary e-mails are welcome. Health and friendship!


| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |


 Breast / Heme (?!)
Breast / Heme (?!)
