Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
verify here. |
Cyberfriends: The help you're looking for is probably here.
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()
 LEARNING OBJECTIVES
LEARNING OBJECTIVES
Once again, consider this all "worth knowing".
Review the liver's architecture and function. Describe its capacity to regenerate, and the limits on this capacity.
Describe the lesions that can produce jaundice. Cite physiology to place them into the appropriate categories.
Use, and furnish (given the definition), each word in the glossary and elsewhere in the handout.
Tell how alcohol affects the liver.
Give a full account of what is known, and what remains unknown, about non-alcoholic steatohepatitis. Tell how to use clinicial data to distinguish this from secret alcohol abuse.
Give a complete account of the generalized syndrome of liver failure, and the causes of massive hepatic necrosis.
Describe various conditions that result in ischemia of the liver. Describe the causes and effects of thrombosis of the hepatic and portal veins.
Describe the pathophysiology and clinical problems seen in portal hypertension.
Describe the viral hepatitis family in substantial detail. Describe the significance of various lab tests and biopsy findings in various stages of these illnesses. Describe the "lupoid hepatitis" family of illnesses, and primary biliary cirrhosis.
Define cirrhosis, and describe its pathophysiology in detail. Describe distinguishing features of each of the many causes of cirrhosis.
Describe cholangitis, and liver abscesses.
Describe the common hepatotoxic agents, and their effects.
Tell how liver failure occurs in children, and what the clinician and pathologist will see.
Describe gallstones and their adverse effects. Describe all the common cancers of the hepatobiliary tree.
Recognize the following gross lesions:
acute yellow atrophy
cavernous hemangioma
cirrhosis (various types)
congestion ("nutmeg liver")
focal nodular hyperplasia
hepatocellular carcinoma
hepar lobatum
Liebermeister grooves
metastases to the liver
Riedel's lobe
Recognize and distinguish the following microscopic lesions:
acute cholecystitis
acute viral hepatitis
alcoholic hepatitis
alpha1-antitrypsin globules (PAS+)
ascending cholangitis
bridging necrosis
bile plugs and lakes
cavernous hemangioma
cholangiocarcinoma / adenocarcinoma of gallbladder
chronic hepatitis
chronic cholecystitis
cirrhosis (generic, and various etiologies)
congestion / central ischemic necrosis
Councilman body
fatty change (microvesicular, macrovesicular)
giant cell ("neonatal") hepatitis
giant mitochondria (PAS-)
ground glass hepatocytes
hepatocellular carcinoma
interface hepatitis ("piecemeal necrosis")
iron overload (1, 2)
liver cell unrest
lobular disarray
Mallory's hyaline
massive necrosis
primary biliary cirrhosis
Wilson's disease
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- Liver / Biliary
Lectures follow Textbook
QUIZBANK: Liver and biliary (all)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ALPHA-1 ANTITRYPSIN (alpha-1 protease inhibitor, a serine protease inhibitor or "serpin"; Am. J. Clin. Path. 139: 184, 2013): A useful protein produced by the liver for the bloodstream. It keeps the body's tissues, notably its elastin, from being totally digested early in life by neutrophil elastase (its principal substrate). If its release from the endoplasmic reticulum is prevented by the mutation (Z-allele which polymerizes), it appears as d-PAS-positive granules of varying sizes within hepatocytes. (* This can happen in advanced chronic liver disease from any cause, but is far more likely an unrecognized antitrypsin abnormality: Am. J. Clin. Path. 107: 692, 1997).
ASTERIXIS: "Liver flap". The tremor of early hepatic encephalopathy.
|

|
BALLOONING DEGENERATION: Hydropic swelling of a hepatocyte (i.e., mild, probably-reversible cell injury). If the cell dies, it is BALLOONING NECROSIS.
BILE ACIDS (BILE SALTS): Sterols that help solubilize bile. From your biochemistry course.
BILE DUCTULE: The little bile ducts at the edges of the portal triads. They feed into the interlobular bile duct. Also called "canals of Hering".
THE BILE DUCTULAR REACTION: Today's preferred name for "ductular proliferation", a marker for extrahepatic bile duct obstruction from any cause. You'll see obvious tubes of cuboidal cells where the canals of Hering should be.
BILE LAKE: An accumulation of bile that has ruptured a canaliculus
BILE PLUG: Bile visible in a distended canaliculus
{12220} jaundice
|
|
BILOMA: A pool of bile in a traumatic (laceration, stab, surgery) lesion of the liver.
BRIDGING NECROSIS: Necrosis linking two portal areas or a portal area and a central area.
CHOLESTATIC JAUNDICE: Jaundice caused primarily by failure of conjugated bilirubin to be sent successfully to the gut
CHRONIC HEPATITIS Morphologic evidence of inflammation AND necrosis plus labs and/or clinical evidence of liver disease for six months or more.
* You'll find pathologists who prefer to call it "chronic necroinflammatory injury".
CHRONIC ACTIVE HEPATITIS: This is an out-of-use term that meant Inflammation + interface hepatitis + fibrosis involving the liver for six months or more. This histologic pattern supposedly meant that the disease would progress to cirrhosis.
CHRONIC PERSISTENT HEPATITIS: This is an out-of-use term for lymphocytes and/or plasma cells in the portal areas, without ongoing necrosis; symptoms and/or abnormal labs for >6 months. This histologic pattern supposedly meant that the disease would not progress to cirrhosis.
The tendency nowadays is to group chronic persistent hepatitis and chronic active hepatitis together as "chronic hepatitis", and not to try hard to distinguish them on morphologic grounds.
* Future pathologists:
Inflamed patches in the sinusoids, away from the interface:
To describe the hepatitis, choose whichever is worse. To use the METVAR algorithm for "histological activity", add the two numbers (0-3 for each), and report the sum, or three if it is more than three.
Stage:
CIRRHOSIS: Scarring of the whole liver sufficient to seriously interfere with proper perfusion of hepatocytes. Instead of the familiar lobules, you'll see fibrous bands dividing the liver into more-or-less round REGENERATIVE NODULES.
|
Australian Pathology Museum High-tech gross photos
|
|
|
CONJUGATED BILIRUBIN: Bilirubin that has been conjugated to glucuronic acid, making it water-soluble
CONFLUENT-LYTIC NECROSIS: Death of clusters of hepatocytes (* attributed in the current literature to humoral immunity)
COUNCILMAN (ACIDOPHIL) BODY: Single-cell necrosis (apoptosis) of a hepatocyte, typically in hepatitis as a result of attack by a T-killer cell.
* CYTOPLASMIC DISSOCIATION means edema at the edges of a hepatocyte, granular cytoplasm around its nucleus. The cell is injured.
FOCAL NECROSIS: Death of individual cells, evidenced either by Councilman bodies or lytic necrosis (i.e., collapse seen on reticulin stain). Inside the lobule, it's "focal lobular necrosis", as in smoldering hepatitis from any cause.
* FEATHERY DEGENERATION: A pattern seen when a liver cell retains both bile salts and water. Ask a physical chemist how the detergent effect of bile salts causes it. When the bile actually digests a group of liver cells, it's called a BILE INFARCT (misnomer, of course). Leave recognizing these to us.
GIANT MITOCHONDRIA (megamitochondria): Monsters seen in hepatocytes in alcoholism (sometimes NASH). They are d-PAS negative (lets you distinguish them from alpha-1 antitrypsin). See J. Clin. Path. 45: 412, 1992.
* "Yokoo bodies." These mitochondria may have suffered a characteristic loss of DNA due to alcohol-induced free-oxygen-radical damage or something; the deletion makes it harder for the liver cell to burn fat, and so forth (Gastroent. 108: 193, 1995.)
GROUND GLASS HEPATOCYTES: Distinctive hepatocytes seen in chronic (not acute) hepatitis B infection. The "ground glass" cytoplasm is an unusual accumulation of a cytokeratin (Hepatology 28: 347, 1998).
HEPATOCELLULAR JAUNDICE: Jaundice due primarily to failure of hepatocytes to properly take up / conjugate bilirubin.
HEMOLYTIC JAUNDICE: Jaundice due to excessive destruction of red cells or their precursors at any site
* HELLP SYNDROME: Hemolysis, elevated liver enzymes, low platelets. A poorly-understood and very serious complication of pregnancy. Seizure and hypertension management, glucocorticoids, and/or exchange transfusions may be required. More about this later.
INTERFACE HEPATITIS: Necrosis of groups of hepatocytes within the limiting plate. The term has replaced "piecemeal necrosis", to prevent confusion with focal necrosis deeper within the lobule. Often the only evidence of "necrosis" that you see is a little area with collapsed architecture (reticulin stain shows the collapse); if you're lucky, you may spot a Councilman body.
INTERLOBULAR BILE DUCT: The big bile duct in the portal tract. It runs with the branch of the hepatic artery.
JAUNDICE: Too much bilirubin (conjugated or not) in the bloodstream, for any reason
{12220} jaundice
LIMITING PLATE: The row of hepatocytes immediately adjacent to the portal tract. It should be smooth and uniform.
* NONSPECIFIC REACTIVE HEPATITIS ("liver cell unrest"): Increased prominence of Kupffer cells and increased ploidy of many hepatocytes. This is a non-specific finding, common to many (if not most) serious illnesses affecting the entire body.
LOBULAR DISARRAY: Loss of the normal radial arrangement of liver plates within the lobule, typically with severe distortion of the sinusoids. The hallmark of acute hepatitis.
LUPOID HEPATITIS: An unfortunate term for the several kinds of non-viral (?), autoimmune hepatitis in which the histology is that of chronic hepatitis, usually with a lot more plasma cells than in the viral forms (worth remembering).
LYTIC NECROSIS: The hepatocytes in a region (large or small) are gone, leaving behind collapsed stroma. Older references call this "dropout necrosis".
MALLORY'S HYALINE: Masses ("rope-like", "cottage cheese", "Mallory-Denk bodies") of altered cytokeratin and cell stress proteins (ubiquitin, others: Arch. Path. Lab. Med. 114: 589, 1990). Usually (but not always) a marker for alcoholic hepatitis / severe NASH.
MASSIVE NECROSIS: Most of the hepatocytes on the slide are dead. Due to poisoning, viruses, medication reactions, or ischemia. SUBMASSIVE NECROSIS means that at least some entire lobules are destroyed, but in other lobules, enough cells are alive.
MACROVESICULAR FAT: One large lipid drop in a hepatocyte, pushing the nucleus to one side
MICROVESICULAR FAT: Several lipid drops in a hepatocyte; the nucleus stays in the center
OBSTRUCTIVE JAUNDICE: Cholestatic jaundice caused by mechanical obstruction of the common bile duct or hepatic ducts. Also called SURGICAL / SURGEON'S JAUNDICE; all other forms of jaundice are MEDICAL / INTERNIST'S JAUNDICE.
* ONCOCYTIC HEPATOCYTES (oxyphilic hepatocytes, i.e., mitochondrion-packed) are common in many livers, especially where there's been a lot of regeneration, i.e., cirrhosis, which has let mutant mitochondria overgrow (Virch. Arch. 432: 349, 1998). Fibrolamellar hepatocellular carcinomas are also mitochondrion-packed.
INTRODUCTION
Is life worth living? It depends on the liver!
-- Anonymous
The liver is usually our heaviest internal organ, and the most durable. Unlike lungs, kidneys, heart, and brain, the livers of most 100-year-olds are morphologically and functionally normal.
Liver pathology includes only a few common diseases. The terminology and morphology of these lesions are notoriously confusing for beginners. Further, you'll have to know them, because liver biopsy is fairly common, especially in this era of managing chronic hepatitis by biopsy results.
It would be best for you to start by learning the definitions in the "Glossary", and making note of the material under "For Future Liver Pathologists".
You already know that the liver is the great chemical plant of the body. You remember its location, its anatomic relationships, its blood supply, and its essential architecture.
Worth mentioning: The STELLATE CELLS ("Ito cells", "perisinusoidal cells") sit in the space of Disse, store vitamin A, and turn on to carry out fibrosis of the hepatic lobule in developing cirrhosis.
* Possbly the most interesting recent work on liver pathology is the finding that signals from hepatocytes in their death-throes stimulate both hepatocyte progenitors and myofibroblsts / stellate cells / laminin producers to handle the re-growth (Gut 59: 645 & 655, 2010) The new hope is that natural killer cells can be activated by biotech products to destroy the proliferating stellate cells (Gut 60: 90, 2011) or at least suppress them (Dig. Dis. Sci. 55: 261, 2010; Gastroenterology 138: 347, 2010).
Normal adult livers weigh 1400-1600 gm. Liver weight is widely variable at autopsy. I've autopsied an end-stage cirrhotic with a 700 gm liver, and an alcoholic with a 7000 gm liver. The liver HURTS when, and only when, its capsule is stretched.
Despite the discussion in "Big Robbins", the normal liver may or may not be palpable, depending on its shape. Maybe 1% of livers have a "Riedel's lobe" easily felt on the right side; this is simply an anatomic variant of no importance to one's health. Others have a small right lobe and a large left lobe, while still others have random grooves across the organ ("hepar lobatum", or one variant). The hyperinflated lungs of the emphysema patient usually push the liver downward and make the edge palpable, but again, this is not reliable; "rib marks" (really from muscle pressure) in emphysema produce the familiar LEIBERMEISTER GROOVES. Remember that a newborn's liver edge is usually easily palpable 1-2 cm below the costal arch.
The histology of the liver is worth reviewing. Remember that the METABOLIC LOBULE ("ACINUS") is centered on the portal areas, and the CLASSICAL LOBULE is centered on the central vein. Whichever system you use, ZONE 1 is the hepatocytes near the portal areas, ZONE 2 is the hepatocytes midway between the portal areas and central veins, and ZONE 3 is the hepatocytes around the central veins.
The familiar polyhedral, pink-staining hepatocytes are often (maybe 10%) binucleate or tetraploid / octoploid. This is normal. You remember the architecture of the liver plates and sinusoids, the passage of bile from canaliculi to canals of Hering to bile ducts, and the appearance and function of the hepatic endothelium and Kupffer cells.
* Prominent Kupffer cells and increased hepatocyte polyploidy is LIVER CELL UNREST, common in people who are sick for a variety of reasons. Its diagnostic significance is nil.
* Future pathologists: You can stain the healthy canaliculi using your CEA stain.
You will learn about biopsying the liver (the open "wedge" biopsy, the classic through-the-skin cutting-neecle techniques, the tiny-pieces transjugular approach for the very-sick) on rotations (Gut 55: 1789, 2006). Lately, a liver cutting-needle biopsy has come to be considered "adequate" if it is 20 mm long and/or contains 11 or more complete portal tracts (Am. J. Clin. Path. 125: 710, 2006).
|
The liver's ability to regenerate is legendary. The Greek titan Prometheus had his liver devoured each day by a monster bird, but it always grew right back.
If individual hepatocytes are destroyed but the architecture of the lobule is NOT destroyed, the remaining hepatocytes will totally regenerate the liver parenchyma. If whole lobules are destroyed, the remaining lobules will expand. They will function normally, though bile may not be drained quite so well. |
 Prometheus |
Of course, if scar tissue alters the flow of blood through the liver (i.e., cirrhosis has occurred), regeneration will only produce less-than-fully-perfused nodules of liver cells. (This will disappoint well-read problem drinkers who understood that their hepatocytes had unlimited capacity to regenerate....)
* Liver biopsies are not always easy to read, especially if the community hospital pathologist isn't focused on liver. The value of a second opinion: Arch. Path. Lab. Med. 125: 736, 2001.
* Incredible as it may seem, your lecturer got his first exposure to pathology as freshman advisee of the dean of experimental liver pathology, Brown's Nelson Fausto, whose focus was and is liver regeneration. ("All right. You say you want to be a doctor. How serious are you?") Turned out to be a fantastic man and teacher. After years of bragging about this, I was delighted to see him as third author of the new "Big Robbins".
Increased bilirubin in the bloodstream is JAUNDICE.
There's no reason to review bilirubin production and metabolism here. You can check "Big Robbins" if you need refreshing.
Here's a simple review, similar to the one in "Big Robbins", of the various causes of jaundice:
TOO MUCH BILIRUBIN BEING PRODUCED ("hemolytic jaundice")
"Ineffective hematopoiesis", i.e., normoblasts dying in the bone marrow
Thalassemias (even mild ones like beta-thal minor)
Megaloblastic anemias
Intravascular hemolysis (many, many reasons for this; IgG-mediated hemolysis of red cells is listed here as well though technically not "intravascular")
Extravascular hemolysis
Big hematomas
GI bleeding
Red infarcts
LIVER FAILS TO TAKE UP AND/OR CONJUGATE BILIRUBIN ("hepatocellular jaundice")
Newborns
Hypoperfusion
Bad alcoholism
Hepatitis (many causes)
Cirrhosis (many causes)
Gilbert's non-disease and the Crigler-Najjar syndromes NOTE: From "Biochemistry". Gilbert's (officially pronounced as French Zheeel-BEAR's) is a forme fruste of Crigler-Najjar (Lancet 346: 314, 1995; Lancet 345: 958, 1995). Both result from mutations of the glucuronyl transferase that solubilizes bilirubin. Crigler-Najjar (two subtypes) is always recessive, Gilbert's can be dominant (interference with normal function) or (more often) recessive (mild loss-of function alleles). Gilbert's is extremely common (several % of the population) and usually a non-problem. One tipoff is that the bilirubin levels, which usually will stay below 6 mg/dL, increase during fasting. Some folks do find it troublesome. Gilbert's may be exacerbated by other illnesses or medications, in particular G6PD deficiency and/or the protease inhibitors used in anti-retroviral therapy (J. Inf. Dis. 192: 1381, 2005). | 
|
LIVER DOESN'T SEND BILIRUBIN TO THE RIGHT PLACE ("cholestatic jaundice")
Problems with the liver cells
Drugs (estrogen, anabolic steroids)
Dubin-Johnson (pigmented) non-disease
* These people lack a pump, which is coded by, of all things, the gene ABCC2 for the hated MRP2 multidrug-resistance protein (Gastroent. 117: 653, 1999) that pumps cancer chemotherapy agents out of cancer cells.
* A specialist can diagnose Dubin-Johnson without biopsy by its effect on different urinary coproporphyrin levels. Don't worry about it.
Rotor (non-pigmented) non-disease.
* Byler's disease ("FIC-1/ATP8B1" and at least two other loci; familial intrahepatic cholestasis). This is a family of autosomal recessive illnesses in which there are problems with the bile transport proteins. There is a problem with bile transport in the liver, and sometimes "Byler bile" is appears coarsely granular. The Byler family from which all the index patients came is Amish and highly inbred; See Hepatology 26: 155, 1997. There is a Byler-like illness at BSEP, the bile salt export pump, and another at the multidrug-resistance protein 3 site).
"Benign familial recurrent intrahepatic cholestasis", the forme-fruste of Byler at FIC-1. Patients have intermittent cholestasis and elevated alkaline phosphatase. On biopsy during an attack, you will see bile in the canaliculi, and only in the canaliculi. Second locus Gastroent. 127: 379, 2004.
Really bad cases of other liver diseases (hepatitis, cirrhosis, alcoholism; i.e., when the liver fails, the picture is likely to be mixed).
Problems with the bile ducts in the liver
Biliary cirrhosis
Biliary atresia
* Alagille's (dysmorphic child, intrahepatic bile ducts in the portal areas vanish over time; autosomal dominant, gene Jagged1 (Circulation 109: 1354, 2004, the variable liver disease itself Gut 49: 431, 2001; molecular biology Am. J. Path. 171: 641, 2007)
Problems with the bile ducts beyond the liver (call a surgeon)
Gallstone in the common duct
Cancer (i.e., biliary, pancreatic, ampullary)
Traumatic / Iatrogenic (i.e., the surgeon nicked the common bile duct)
Note that in all but hemolytic jaundice, bile production will be diminished. Stools may become light-colored (gray if the bile is completely obstructed), and there will be diminished intestinal absorption of fat (pee-yew!) and fat-soluble vitamins.
Lab tests are of considerable help in distinguishing these entities.
Obviously, in the first two categories, the serum unconjugated bilirubin will be elevated.
In the third category, only the conjugated bilirubin will be elevated until the liver cells themselves are damaged. Serum BILE ACIDS ("bile salts") will also be increased from the onset, producing the troublesome itching seen in these syndromes. Conjugated (but not unconjugated) bilirubin in the bloodstream spills into the urine. You'll study other markers for cholestasis in the unit on lab testing.
| Future clinicians: Try grapefruit juice for pruritus of liver disease (Ann. Int. Med, 126: 920, 1997). |
 |
On biopsy, obstructive jaundice presents the familiar BILE PLUGS, which begin as dilatations of the canaliculi and end up forming BILE LAKES when the canaliculi rupture.
* As the liver cells become damaged, they fill with soap bubbles (i.e., bile salts and water), producing FEATHERY DEGENERATION. You won't need to recognize this. Later, you'll see necrotic cells surrounding bile lakes.
WHEN THE LIVER FAILS
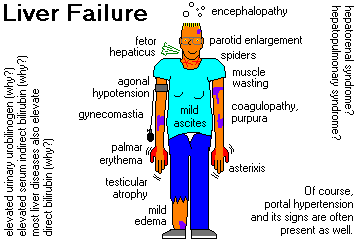
Regardless of cause, when the liver can no longer function as chemical plant, several unwholesome things happen.
JAUNDICE is usual. When the liver is really scrambled, hyperbilirubinemia is mostly the conjugated sort, i.e., the cells remember how to conjugate, but not what to do with, the bile. There is usually some unconjugated hyperbilirubinemia, too.
HYPOALBUMINEMIA is usual, since the liver isn't making albumin. Without albumin in the bloodstream, ascites and edema develop. By the way, HYPOCHOLESTEROLEMIA is usual in liver disease too (unless the primary problem is obstruction of bile flow -- why?), since the liver isn't producing LDL's. (This is part of the reason for the silly myth that "too low cholesterol is bad for you".)
COAGULOPATHY of liver disease (NEJM 365: 147, 2011) results from diminished hepatic synthesis of factors II, V, VII (first to go), IX, and X. (Note that absent vitamin K from malabsorption also prevents synthesis of II, VII, IX, and X.) Monitor all this by following the prothrombin times (rather than PTT, since factor VII is first to go and to return).
The anti-clotting factors are also diminished, and people talk about the clotting system in liver failure being "rebalanced".
Further, as the liver fails to clear factors that have become activated in the course of living, low-grade DIC is likely to develop. This is probably why PT and the classic measures correlate poorly with intractability of bleeding from varices / bleeding during liver transplantation.
As liver cells fail, detoxification of nasty compounds fails and HYPERAMMONEMIA and FETOR HEPATICUS (a distinctive mercaptan-based odor to the breath). Other side-effects are reddening of the thenar and hypothenar eminences ("palmar erythema"), spider "angiomas" (you'll learn about these in physical diagnosis), and (in men) gynecomastia and testicular atrophy. In longstanding liver failure, the parotid glands often enlarge for some reason (still completely unknown as of 2014).
THROMBOCYTOPENIA is due to lack of thrombopoietin: Am. J. Gast. 94: 1918, 1999.
HEPATORENAL SYNDROME is a syndrome of kidney failure.
We used to precipitate this by "lasixing" cirrhotics with ascites.
The pathophysiology, once obscure, is now clear. First, liver failure interferes with the breakdown of the vasodilator nitric oxide. Second, portal hypertension itself forces the splanchnic arteries to open wider at the expense of circulation to the rest of the body. Third, bacteria from the gut find their way into the mesenteric lymph nodes, where they cause all sorts of havoc with cytokines (NEJM 361: 1279, 2009). We now manage all but the worst cases by giving plasma expanders and vasopressin analogues (Gastroent. 122: 923, 2002) (to constrict the splanchnic circulation) plus dopamine (to open the renal microcirculation) helps (Hepatology 27: 35, 1998; Am. J. Gastroent. 92: 2113, 1997; Clin. Sci. 92: 433, 1997; Mayo. Clin. Proc. 71: 874, 1996; Lancet 362: 1819, 2003). When there is massive tense ascites, tapping and draining it ("large volume paracentesis") is helpful in the short-run. Unless the liver disease is reversible (i.e., alcoholic hepatitis or a drug allergy), this is just buying time while waiting for a liver transplant. Update South. Med. J. 103: 654, 2010.
For splitters:
HEPATOPULMONARY SYNDROME is seen when the liver fails. The small arterioles and capillaries of the lungs dilate preposterously causing V/Q mismatching (i.e., the oxygen cannot reach the centers of the vessels, and there are some shunts opening that bypass the alveoli altogether). There is no current remedy apart from curing the liver disease. See Gastroent 113: 606, 1997; Surg. Clin. N.A. 79: 23, 1999; Mayo Clin. Proc. 79: 42, 2004; Lancet 363: 1461, 2004 ("notoriously underdiagnosed"); NEJM 358: 2378, 2008; Med. Clin. N.A. 93: 871, 2009.
* Future clinicians: In contrast to congestive heart failure, dyspnea in hepatopulmonary syndrome improves when lying flat ("platypnea"), since the V/Q mismatching is worst in the lung bases. Any idea why?
* Future clinicians: PORTOPULMONARY HYPERTENSION is the other lung problem caused by liver disease, and often coexists with hepatopulmonary syndrome. It's probably mediated by factors that aren't being cleared from the blood, and the histopathology is as for idiopathic pulmonary hypertension. Patients must have portal hypertension and/or bad liver disease, plus mean pulmonary arterial pressure above 25 mmHg at rest and a few other criteria. Update Hosp. Pract. 41(2): 62, April 2013.
HEPATIC ENCEPHALOPATHY is not a pretty sight, and probably results from a combination of factors, including nitrogen-containing false neurotransmitters (supposedly including octopamine -- remember that from "Biochemistry"? -- and some others) produced by the gut flora.
* Fatigue in liver failure may respond to ondansetron: Lancet 354: 397, 1999. The antibiotic rifaximin seems to help (NEJM 362: 1071, 2010).
Early in the process, there's a curious distortion of spatial perception. (The stereotype of accelerated confusion in the problem drinker is all too familiar -- he pours the whiskey onto his lap, rather than into the glass in his other hand; he cannot find his way home even when he sobers up. Whatever the cause, hepatic encephalopathy makes life far more difficult.) The first change on physical exam is ASTERIXIS, a curious flappy falling-asleep-and-waking-back-up of the fingers-hands-arms-whole body. Clinicians monitor hepatic encephalopathy by measuring blood ammonia.
{01383} Alzheimer II glia in hepatic encephalopathy (best one is in the center of the field; it appears as a swollen, pale nucleus)
In acute massive liver failure, cerebral edema is the pathway out of life in about 50% of cases (Lancet 351: 719, 1998). We're still making educated guesses about the mechanism.
When the liver finally gives up completely, REFRACTORY HYPOTENSION supervenes from total-body vascular relaxation (which we can suppose is due to the failure of the liver to metabolize some vasodilator, most likely one that's not yet been discovered.) Nothing you can do will save the patient.
Reminder: Serum liver enzyme (transaminases, lactate dehydrogenase) concentrations become elevated when liver cells are acutely injured. Note that in burned-out cirrhosis when drinking is stopped, liver enzymes will be normal.
CIRRHOSIS
Cirrhosis ("roaches of the liver", etc.) is scarring of the whole liver sufficient to permanently interfere with circulation of blood to the hepatocytes, no matter what the cause. You will see
NOTE: The development of fibrosis is still quite mysterious; we know the stellate cells are the ones responsible, but nobody really understands it or what we might do to stop it. Sometimes, you can see layer upon layer of reticulin fibers being laid down as liver cells die in waves; this is the sign of irreversible (?) damage in chronic hepatitis, and probably is how scars build up, at least in part.
{00005} cirrhosis
{08846} cirrhosis, trichrome stain (fibrous tissue is blue, of course);
micronodular
{39710} cirrhosis after hepatitis, gross photo showing uneven involvement of the liver lobules.
Just recognize cirrhosis.
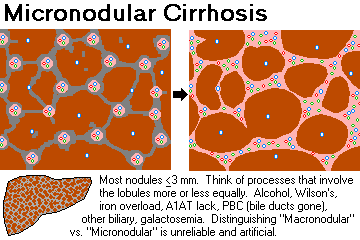
MICRONODULAR CIRRHOSIS: Most of the nodules are smaller than 0.3 cm, and the fibrous-scar bands are relatively thin.
Think of alcoholism, hemochromatosis (since alcohol and iron will involve all lobules equally), primary-autoimmune biliary cirrhosis (since portal areas tend to link to adjacent portal areas), or biliary infection/obstruction (same reason, "secondary biliary cirrhosis"; remember cystic fibrosis).
* The inborn errors of metabolism worth remembering are the bad kind of galactosemia, tyrosenemia, type IV glycogen storage disease, and hereditary fructose intolerance.
{08285} micronodular cirrhosis (this happens to have been a case of primary biliary cirrhosis); liver on left is normal
|
|
|
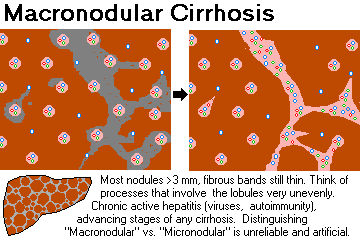
MACRONODULAR CIRRHOSIS: Most of the nodules are larger than 0.3 cm, and the fibrous-scar bands are relatively thin.
Think of chronic hepatitis, with its uneven pattern of inflammation, progressed to cirrhosis (since viral disease is generally patchy and will not involve all lobules equally).
Wilson's disease, galactosemia, and alpha-1 antitrypsin deficiency may supposedly produce either pattern, though they probably begin involving lobules evenly; since they are accumulations and cell death releases the storage product, perhaps this explains the unevenness. As a matter of fact, a rehabilitated alcoholic's micronodular liver will, after a few years of sobriety, exhibit enough large regenerative nodules to qualify as macronodular.
* Pathologists only: "Incomplete septal cirrhosis" is stabilized (regressing?) macronodular cirrhosis with only thin fibrous bands and really no nodules. Liver function tests are better, but portal hypertension may be is more severe -- hence the tendency to say "cirrhosis" despite no nodules. See Gastroent. 106: 459, 1994.
* In an era in which regression of fibrosis is now well-recognized, just how useful the term "cirrhosis" is might be questioned (Am. J. Clin. Path. 137: 5, 2012). Your lecturer believes that the appearance of a cirrhotic liver is so distinctive that the term will always be with us. Please do remember that a diagnosis of "cirrhosis" is no longer a death sentence.
{08441} macronodular cirrhosis
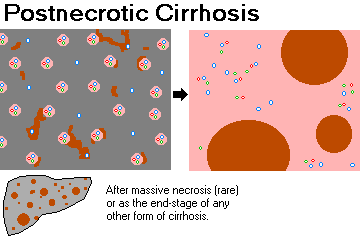
POST-NECROTIC CIRRHOSIS ("end-stage liver"): Macronodular cirrhosis with really big, thick fibrous-scar bands. Usually results either from submassive necrosis (i.e., whole lobules were destroyed), or (much more often) progression of another type of cirrhosis to the end stage (micronodular and macronodular no long mean anything as nodules coalesce).
{25659} macronodular cirrhosis (some big scars show progression to postnecrotic cirrhosis)
Especially in kids cured of thalassemia by marrow transplantation, extensive reversal of cirrhosis is now known to take place (Ann. Int. Med. 136: 667, 2002); there is often some regression when hepatitis C virus is eradicated from a long-time patient (Dig. Dis. Sci. 43: 2573, 1998; Ann. Int. Med. 149: 399, 2008); the new treatments for chronic hepatitis B cause a majority of patients with cirrhosis to revert to non-cirrhosis on histology (Lancet 381: 468, 2013), and when autoimmune hepatitis is controlled (Ann. Int. Med. 127: 981, 1997)
There are now animal models for stem cell work in partially-repairing the damaged liver (as is already in use for the damaged heart). Stay tuned: Gastroent. 135: 438, 2008.
* Death rates from cirrhosis (age-corrected) have run a curious pattern over the past 100 years. Between 1900 and 1934, deaths dropped by about 2/3; this coincided with the temperance movement and the massive decline in alcohol consumption. The end of Prohibition and the Great Depression resulted in a tremendous resurgence of alcohol overindulgence, and the rate of death from cirrhosis skyrocketed, peaking in 1970. Since then, they've dropped dramatically; I suspect the explanation is better nutrition and the recovery movement (Postgrad. Med. 115: 13, Jan 2004.)
CIRCULATORY PROBLEMS
|
CONGESTION of the liver receives excessive attention. There's no mystery; if the right side of the heart isn't pumping well enough, blood pools in the liver. Except in the most sudden violent deaths, the central areas of the liver will be more or less congested. (If you're at an autopsy and someone asks, "Is that a nutmeg liver?", you can safely guess "Yes!") Clinicians enjoy showing the hepatojugular reflux of those with congested livers, especially behind failing right ventricles. Pathologists enjoy exhibiting their cut nutmegs, which have light-and-dark areas that resemble congested liver. * If you want to get fancy... instead of demonstrating hepatojugular reflux, you can now use a high-tech device to assess the "stiffness" of the liver, which is of course increased in the living when they have congestive heart failure in the absence of co-existing cirrhosis (Radiology 257: 872, 2010). |  |
{03949} nutmeg liver
{31889} nutmeg, real
|
|
If death has been preceded by a few hours of inadequate circulation (heart failure, shock), count on seeing some hepatocyte necrosis in the centers of lobules. (This is CENTRAL HEMORRHAGIC NECROSIS. Why the liver? Why in the centers? Think about it!)
This isn't "due to the congestion", but merely results from inadequate perfusion with oxygenated blood.
Clinicians may have noted "elevated liver transaminases" ("ISCHEMIC HEPATITIS"), and even mild jaundice. You can experience the transaminase elevations yourself by running a marathon. Don't worry, the liver will completely regenerate (since its connective tissue framework is still intact.)
If hepatic congestion and underperfusion have been extreme and longstanding, the rare CARDIAC SCLEROSIS may supervene. This is substantial fibrosis in the central areas of the lobule. (Grossly, the liver surface looks like a football, since scar contracts in the centers of the lobules.)
In extreme cases (i.e., tricuspid insufficiency, certain congenital heart surgeries), the fibrous tissue may bridge adjoining lobules -- true CARIAC CIRRHOSIS.
* That cardiac cirrhosis is real has recently been demonstrated by a study of people who have undergone the Fontan procedure, in which the right ventriclar pulse is transmitted directly to the hepatic veins. See J. Thorac. Card. Surg. 129: 1348, 2005.
* In cardiac cirrhosis, the central veins may become connected to one another, leaving "reverse lobulation", each with a single portal tract in the middle.
Otherwise, cardiac sclerosis is usually just an anatomic pathologist's curiosity.
LIVER INFARCTS
The liver has a dual blood supply and, while hepatocytes are vulnerable to hypoxia, the stroma is very resistant and hepatocytes regenerate easily. This makes it difficult to truly arterially infarct the liver.
When a branch of the portal vein is compromised, the worst that usually happens is atrophy of hepatocytes in a region ("Zahn's infarct"; fresh lesions have much stasis of blood in the sinusoids and thus look blue).
HEPATIC VEIN THROMBOSIS ("Budd-Chiari")
{49262} Budd-Chiari; liver is engorged with blood and you can see the clots;
Sounds serious, and is. The most common cause is polycythemia vera. Most any other cause of hypercoagulable blood can produce "Budd-Chiari". Another important cause is invasion of the hepatic veins by hepatocellular carcinoma.
As you'd expect, in the acute case, the liver swells (ouch!), ascites develops rapidly, and the patient usually dies of venous infarction of the liver unless surgery or thrombolysis are performed.
In some foreign countries, "chronic Budd-Chiari" is a common problem. Nobody knows why. At autopsy, look for fibrous "webs" in the hepatic veins.
* Diabetic micro-angiopathy occasionally produces non-cirrhotic fibrosis (like hyaline arteriolar sclerosis) of the sinusoids. The entity is newly-named "diabetic hepatosclerosis" (Arch. Path. Lab. Med. 130: 27, 2006; update Am. J. Clin. Path. 132: 494, 2009). Probably it is common but under-recognized. Ordinarily the hepatic sinusoids have no basement membrane; if you see one, it's probably diabetic hepatosclerosis.
HEPATIC VENO-OCCLUSIVE DISEASE, clinically a Budd-Chiari mimic but with no thrombus, results from intimal thickening of the veins (onion-skinning, etc.). Think of Jamaican bush-tea (as in the lung: terrible health problem West. Ind. Med. J. 64: 60, 1997), comfrey, graft-vs.-host, radiation effect. Older review, emphasizing the "herbal tea" connection: Arch. Surg. 125: 525, 1990). Several high-power medications (sirolimus, thioguanine, oxaliplatin) have probably caused veno-occlusive disease. And it's one of the most serious complications of hematopoietic stem-cell transplantation (Lancet 379: 1301, 2012 -- * defibronate study).
![]() Vaso-occlusive disease
Vaso-occlusive disease
Joel K. Greenson MD
U. of Michigan
* Sickle-cell patients often have chronic venous outflow obstruction (why?). Be careful about biopsying these people. Blood 101: 101, 2003.
PORTAL VEIN THROMBOSIS
Again, this is serious. It results from hypercoagulable blood, invasion by hepatocellular carcinoma, pancreatitis, or cirrhosis.
The major problems are ascites and venous infarction of the bowel.
NECROSIS OF THE LIVER
Infections tend to produce random areas of necrosis ("focal", "spotty"), ranging from tiny (most cases of viral hepatitis) to massive ("acute yellow atrophy" / "acute liver failure").
Poisons and other noxious things, on the other hand, tend to damage distinctive portions of the lobule (why?) More about this later.
CENTRAL NECROSIS: Ischemia, carbon tetrachloride, chloroform, acetaminophen
{07020} carbon tetrachloride, gross; note the necrosis (yellow, of course)
{07022} carbon tetrachloride, microscopic
MIDZONAL NECROSIS: Yellow fever and dengue. (Think about why. It is in the virus's interest to have regenerating hepatocytes on both sides.)
PERIPHERAL NECROSIS: Acute iron poisoning (J. Tox. 39: 721, 2001), phosphorus, eclampsia/HELLP (in eclampsia/HELLP, fibrin microthrombi should be visible in the sinusoids near the portal areas).
{07023} liver showing phosphorus poisoning; note periportal necrosis
PELIOSIS HEPATIS ("blood cysts", a misnomer)
Lakes of blood among the hepatocytes. Usually a non-problem found on imaging or autopsy. On section, the liver features many easily visible holes filled with blood.
The pathology has recently been reviewed in depth (For. Sci. Int. 149: 25, 2005.)
In the more familiar sort of peliosis the lakes are lined only by hepatocytes ("parenchymal peliosis"); in this case, the lesions are irregularly-shaped.
Anabolic steroid use is the best-known cause (generally parenchymal), but many others are known (oral contraceptives, cachexia) or suspected (hemangiomas, congestion in people with mild weakness of the veins).
A blow to (or biopsy of) the involved organ may cause these to rupture, with serious hemorrhage.
INFECTIONS
VIRAL HEPATITIS: GENERAL CONSIDERATIONS
The hepatitis family is an alphabet-soup of viruses. However, the anatomic pathology is generally similar. Some viruses are better at producing different patterns than are others.
You can get each of these infections only once. But B can linger, and C usually does linger, as a minor or major problem.
As with most viral diseases, infectivity peaks just before symptoms appear. Acute hepatitis is heralded by the blahs. As the immune system gears up, joint pains and rash can occur. Appetite vanishes, and the patient typically becomes utterly revolted by tobacco. (Smoking cessation is a redeeming feature of the acute hepatitis family.)
In the acute disease, the liver swells and becomes tender, jaundice often appears (mild cases are "anicteric"), and (with influx of bile into the bloodstream) the patient starts to itch and to pass brown urine (why?) Serum transaminases go sky-high, and other lab evidence of liver disease may become apparent.
The best treatment your lecturer knows for the acute phase is masterful inactivity for all but C, intensive therapy for C. Educate the patient, find out who else needs to be checked for hepatitis or get prophylactic gamma globulin, and give clotting factors if you must.
ACUTE VIRAL HEPATITIS: You will see
Note that in hepatitis, the cells may die either by lysis or apoptosis, or both. Probably the lysis is due to the viruses and/or to antibodies, while the apoptosis is caused by instructions to self-destruct delivered by the T-cells.
{05961} acute viral hepatitis with Councilman body
{08834} acute viral hepatitis; sinusoids are
not visible, lots of inflammatory cells
{11787} acute viral hepatitis, great bile plugging
{12764} acute viral hepatitis
{12758} acute viral hepatitis
{12767} acute viral hepatitis
{12770} acute viral hepatitis (do you see a Councilman body?)
{12773} acute viral hepatitis
{12776} acute viral hepatitis
|
|
|
|
|
|
MASSIVE NECROSIS ("FULMINANT HEPATITIS"; "ACUTE YELLOW ATROPHY") may supervene on any kind of acute hepatitis, and often kills the patient in short order. Update Lancet 376: 190, 2010.
Causes range from medicines (idiosyndratic responses to real medicines; adulterants in quack medicines, really bad hepatitis B, D, or E; mushroom poisoning; rare complications of pregnancy; the worst cases of typhoid; extreme vascular disease; rarely hepatitis A, maybe not hepatitis C)
Grossly, as you would expect, the liver is shrunken, red-to-yellow, soft, and flabby, with a wrinkled capsule.
Histologically, the hepatocytes are almost all gone (lytic necrosis and/or apoptosis), leaving a collapsed fibrous tissue framework. Don't expect to see much inflammation.
SUB-MASSIVE NECROSIS is a little less striking histologically and lasts a little longer, killing the patient in a few months. (Or the patient may recover after being super-sick for a few months.)
If a patient survives either process, the parenchyma is usually intact, and recovery should be complete, without cirrhosis. Rarely, the collapsed reticulin meshwork of the liver turns into broad scars (instant "post-necrotic cirrhosis").
{13320} massive necrosis after hepatitis, gross (nothing left but the reticulin and endothelial framework!)
{13321} massive necrosis after hepatitis, histology
{13322} massive necrosis after hepatitis, histology
CHRONIC HEPATITIS: Inflammation of the liver for more than six months.
You will see a dense, mostly-lymphocytic inflammatory infiltrate in the portal areas, with or without spill-over into the parenchyma.
There may be some smoldering changes resembling acute hepatitis in the parenchyma.
In mild cases, the limiting plate is intact (i.e., there is no interface hepatitis). We used to call this "chronic persistent hepatitis".
{12779} mild chronic hepatitis, story
{12785} mild chronic hepatitis, H&E (not a very good case, since lymphocytes are very few, and a
portal
area is not even shown; indistinguishable from mild acute hepatitis)
{12788} mild chronic hepatitis, reticulin stain (see the limiting plate intact);
you don't need to tell this isn't normal liver;
{12791} mild chronic hepatitis, trichrome (again, see the limiting plate intact);
again, you can't tell this isn't normal liver;
These findings are more ominous when the patient has chronic hepatitis clinically:
Drug-induced liver disease (most notably methotrexate), Wilson's, alpha-1 antitrypsin deficiency, and the autoimmune "lupoid" hepatitis family also typically pass through a "chronic active hepatitis" histopathology stage on their way to cirrhosis.
{12800} severe chronic hepatitis, piecemeal necrosis
{12803} severe chronic hepatitis; subtle
{20328} severe chronic hepatitis; almost to cirrhosis (the
nodules are not yet completely separate)
{40279} severe chronic hepatitis, note the necrotic cells
{20183} severe chronic hepatitis with good bridging necrosis;
the hepatocytes are stained orange and the bridge is an area of lytic necrosis
|
|
![]() Hepatitis C
Hepatitis C
Joel K. Greenson MD
U. of Michigan
Future pathologists please note: Any and all of these patterns (from acute hepatitis to post-necrotic cirrhosis) can be mimicked by idiosyncratic reactions to various drugs. Chronic hepatitis and its sequelae are often caused by autoimmunity.
HEPATITIS A ("infectious hepatitis")
This is an unpleasant but almost always self-limited disease caused by a tiny RNA enterovirus (* picornavirus; "pico-" means "tiny", and "rna" you can figure out).
{00444} hepatitis A virus
{08173} hepatitis A virus
Hepatitis A is transmitted by the fecal-oral route, i.e., poor sanitation, small kids (i.e., day-care or institutions), hands (J. Clin. Micro. 30: 757, 1992, note that there are countries where toilet paper isn't used), raw oysters (be sure to ask), some gay male sexual practices (JAMA 267: 1587, 1992), others. Hepatitis A is more common overseas but is no rarity in the U.S.
Hepatitis A, until recently an endemic scourge on many Indian reservations (MMWR 46: 600, 1997) has become much less common thanks to the recommendation to immunize all these children (Am. J. Pub. Health 94: 996, 2004).
* The 1998 strawberry outbreak: NEJM 340: 595, 1999.
The incubation period is about two weeks, and this is the time when virus is shed in the feces. The infection in kids is usually asymptomatic. Adults who get symptoms at all suffer jaundice and discomfort for a few weeks. Once in a while, the disease is fatal.
You'll hear different versions of whether the virus itself damages hepatocytes (the other enteroviruses are cytotoxic), or whether the liver damage is actually wrought by the body's immune response.
Immune response is exactly what you'd expect:
IgM ANTI-HAV appears in the blood when the symptoms begin, clears the infection (which may wax and wane clinically for a few months), and usually disappears within 12 months.
IgG ANTI-HAV appears in the blood during the symptomatic period, and usually stays around for life, rendering the patient immune.
Occasionally the disease causes acute yellow atrophy and death/transplantation (Am. J. Gastro. 98: 448, 2003).
Hepatitis A very seldom becomes chronic or leads directly to cirrhosis. There is probably no carrier state. At worst, the disease might be a trigger for autoimmune chronic hepatitis, but the virus won't stay around.
* How the vaccine came about: Lancet 343: 321 & 322, 1994; J. Inf. Dis. 169: 996, 1994; JAMA 271: 1328, 1994; JAMA 273: 906, 1995 (now classic). Hepatitis A vaccine for post-exposure prophylaxis seems to work about as well as immune globulin: NEJM 357: 1685, 2007.
HEPATITIS B ("serum hepatitis"; update NEJM 359: 1486, 2008; Lancet 373: 582, 2009)
The world's most serious DNA-virus-related health problem. The reservoir for the virus ("HBV", "Dane particle") is the world's 300 million (Proc. Nat. Acad. Sci. 93: 6542, 1996) or 350 million (WHO 2009) or 400 million (ASCP) carriers, most of whom are asymptomatic and probably have histologically normal or near-normal livers.
{00445} hepatitis B virus
{08175} hepatitis B virus
{10532} hepatitis B, * orcein stain (stains the virus)
{11708} hepatitis B, core antigen stained in nuclei
Even an infinitesimal amount of infected blood, when introduced into another person's tissues, is highly effective in transmitting the infection.
Routes include
People born uninfected in the poor nations also frequently turn positive during their childhood. This has been blamed on bedbugs; probably this isn't the main problem (Lancet 343: 761, 1994).
In the U.S. underclass, infection is also rampant, with around 25% of forensic-service deaths being core-antibody positive (J. For. Sci. 38: 1075, 1993).
* Hepatitis B immunization has resulted in a triumphant reduction in the prevalence of carriage in Taiwan and the rate of hepatocellular carcinomas (JAMA 276: 906, 1996).
* In 1997, I predicted the 1998-9 media hype that the vaccine causes multiple sclerosis. The activists don't have the numbers, but even after the claim was already totally discredited, many lawsuits still got filed (Science 281: 630, 1998).
* Catching hepatitis B from the surgeon, even when e-antigen-negative ("low-risk"): NEJM 336: 178, 1997.
Virus antigens:
HBsAg ("Australia antigen"): Surface antigen. Envelope protein. During the productive infection, the liver cells make considerable excess non-infectious HBsAg, facilitating diagnosis.
HBcAg: Core antigen. Nucleocapsid.
HBeAg: Another nucleocapsid antigen, which means the virus is being replicated.
* Interestingly, entry of the virus into the hepatocyte is by means of binding to polymerized serum albumin.
After a person first meets the virus, the incubation period is usually 1-4 months.
Antigens and antibodies:
HBsAg first appears in the blood shortly before symptoms begin (if they are to begin -- maybe 70% of infections are asymptomatic). It remains in the blood for the duration of the infection, whether it is acutely symptomatic, slowly-progressive / subclinical, or merely the carrier state.
HBeAg appears in the blood just after HBsAg, and before symptoms start. It remains as long as there is acute viral replication (you're VERY contagious....), and disappears if (and only if) viral replication stops. The patient is still sick when HBeAg disappears, but can take comfort in the good news.
Anti-HBeAg appears soon after viral replication and HBeAg production stop (if they stop). The patient can still be sick, but this is another piece of good news.
If HBeAb stays around, it's a marker for the person being very contagious -- bad enough that the NCAA forbids sports participation for a chronic carrier.
Don't ask for an assay of HBcAg, the core antigen in the blood. It's an intranuclear protein and there's almost none in the blood. However, Anti-HBcAg, in its IgM form, appears in the blood typically before symptoms begin, and generally remains present for years (IgG anti-HBcAg will eventually take over, maybe). If a person with clinical hepatitis has cleared his blood of HBsAg, but has not yet developed detectable anti-HBsAg, the presence of IgM anti-HBcAg confirms that the infection is, indeed, hepatitis B and is in the CORE WINDOW.
Anti-HBsAg is generally present when the infection is pretty much over, and is a good sign of recovery.
NOTE: Now that we use viral DNA to discover hepatitis B infection, we're discovering quite a few folks who never mount an antibody response. These folks seem to fail to expand their clone of HBV-specific T-cells (Gastroent 134: 1470, 2008).
"Occult hepatitis B" has undetectable HBsAg, positve anti-HBc, and positive anti-HBs, but there's sometimes viral DNA detectable in the blood, and the virus is still hiding in the liver. Its true frequency is of course unknown. These people will have the disease flare when they get immunosuppressed by chemotherapy, infamously by rituximab. Overall, about 1/3 of patients who are HBsAg-positive will reactivate (i.e., the virus reappears in the blood), and of these, maybe a quarter will get very sick, especially if they were not given lamivudine to prevent it. People with occult infection (i.e, anti-HBs negative, anti-HBc positive) may serorevert (HBsAg reappears in the blood) during chemotherapy.
BEWARE! During the time between disappearance of HBsAg and appearance of anti-HBsAg, the patient may experience a potentially-lethal type III systemic vasculitis. (Why?)
If your patient is anti-HBsAg positive and anti-HBcAg negative, probably this person has had the hepatitis B vaccine (why?)
* Well, maybe it's not a SURE sign of recovery; the Scripps crew has found viral DNA up to five years after appearance of the antibody, but the patients don't seem sick or catching (J. Clin. Inv. 93: 230, 1994).
Symptoms begin in hepatitis B infection only when T-cells become angry with HBsAg and HBcAg and start killing the hepatocytes that produce them. Histopathologists find T-cytotoxic cells where the hepatocytes are dying. Eventually, the only surviving liver cells are the ones that won't continue making viruses, and these replenish the liver.
The acute disease may be subclinical, or can cause weeks of jaundice and misery, or can cause fulminant hepatitis and death (this used to kill a few percent of patients with acute hepatitis B before we had effective antiviral medications), or sub-massive hepatic necrosis with resolution or cirrhosis.
Survivors (and 99% of people survive the acute episode) usually clear themselves of the virus, but maybe 10% fail to do so. These can become healthy carriers, develop chronic hepatitis that may remit or progress to cirrhosis if untreated. Rule of thumb: The more severe the initial illness, the less chance of remaining chronically infected (why?) Terminology: Chronic hepatitis B means HBsAg has been present in the bloodstream for 6 months or more.
NOTE: Carrying hepatitis B, with or without ongoing liver disease, is an important cause of cryoglobulinemia and/or "polyarteritis nodosa of hepatitis B" (both immune complex, type III immune injury problems).
NOTE: People who become carriers are those who mount a poor immune response. Men (weaker immune response) are more at risk than women; different HLA types differ in susceptibility (Lancet 344: 1194, 1994).
NOTE: Quite a few patients with hepatitis B in the blood but persistently normal transaminases do indeed go on to fibrosis and cirrhosis (Gastroent. 134: 1376, 2008).
Further, anyone who carries around the virus for a long time is at substantial risk for hepatocellular carcinoma. (Hepatitis B and/or hepatitis C contribute to most cases of this cancer, which worldwide is one of the most common fatal diseases. In the case of hepatitis B, the virus may be acting as a mitogen that allows Nowell's law to act, and/or mutating genes at or near its insertion sites: J. Virol. 65: 6761, 1991; there is no doubt that insertion of the virus can and does scramble chromosomes: Proc. Nat. Acad. Sci. 88: 9248, 1991.)
People who continue to harbor the virus are probably those that are not especially good at making interferon (the chronically sick, the immunocompromised, little kids, the unlucky, men much more often than women). Alpha-interferon was the original the mainstay of therapy for chronic hepatitis B infections, and the results are encouraging, with maybe half of people going into remission. Of course, interferon therapy was expensive and produces 'flu-like symptoms, but it was better than dying or infecting your spouse. And thankfully the risk for hepatocellular carcinoma also dropped greatly (Cancer 66: 2395, 1990 was the first big one).
Future histopathologists: You can stain for HBsAg in the cytoplasm, or core antigen in the nucleus. "GROUND GLASS HEPATOCYTES", with greatly increased smooth endoplasmic reticulum suggest chronic hepatitis B infection.
* Don't worry about the "sanded nuclei", full of core antigen, that are classically described in hepatitis B. This is subtle and of no use clinically, and we won't ask you to recognize them.
We may hope that the hepatitis B vaccine will eventually make this infection, and its dread sequelae, a thing of the past. Gambia institutes HBV vaccination of its population (Lancet 341: 1129, 1993). Kids in the U.S. should get immunized, too (Pediatrics 93: 747, 1994). Please be sure you, too, are immune, Doc.
HEPATITIS D (Lancet 378: 73, 2011)
"Delta hepatitis virus" (HDV) is an incomplete RNA virus that can replicate only while synthesis of HBsAg is also taking place. Unlike HBV, delta is directly cytopathic to hepatocytes. About 15 million people are infected worldwide.
Delta may CO-INFECT (i.e., arrive under a person's skin at the same time as the HBV particle) or SUPERINFECT (i.e., arrive under the skin of a person already infected with HBV). Fortunately, delta is relatively hard to transmit (somewhere between HBV and HIV in infectivity), and hepatitis D is most common in gay men and (in the developed world, especially ) IV-drug-abusers.
The results are grim. In co-infections, fulminant disease is common (maybe 5%). In a superinfection, the victim experiences a second round of acute hepatitis, which tends (maybe 50% of the time) to turn chronic and progressive. Treating chronic hepatitis D with alpha-IF: NEJM 330: 88, 1994 (it helps around half of them while being treated; half of these relapse.) If the virus continues replicating, death is near-certain after a few decades (Gastroenterology 136: 1629, 2009).
Fortunately, carriers of delta are probably uncommon. Delta kills maybe 1000 people a year in the USA.
* Disturbingly, there's a fair amount of hepatitis D in school children in Mongolia, and it seems to be acquired through dirty needles in the health care setting (Am. J. Trop. Med. Hyg. 75: 365, 2006).
* LABREA HEPATITIS, noted in the western parts of the Amazon region, is a fulminant, deadly hepatitis of children and young adults caused by hepatitis B and D. There is microvesicular steatosis, and inflammation of the portal and central veins, and "morula cells", macrophages loaded with virus (Trans. Royal Soc. Trop. Med. 101: 831, 2007).
HEPATITIS C (the vast majority of the old non-A, non-B hepatitis cases) updates Ann. Int. Med. 132: 296, 2000; Mayo Clin. Proc. 73: 355, 1998; Lancet 362: 2095, 2003.
This flavivirus (HCV) and its related disease spectrum are now well-characterized. Worldwide, about 170 million people carry the virus (Gut 58: 846, 2009). In the U.S., 1% of asymptomatic people are positive for HCV (maybe more than this among swingers and MUCH more among IV drug users; maybe 0.3% in those not in these risk groups; health care workers aren't at increased risk: Lancet 343: 1618, 1994; ear-piercing is a risk factor: NEJM 334: 1691, 1996; 19% positive for inner-city forensic-pathology service deaths J. For. Sci. 38: 1075, 1993); in the 2000's, intravenous drug use was the most common cause in the USA, with 98% of junkies positive in some communities; in the poor nations, it's around 5%; worldwide 3%; the highest known prevalence is around 20% in Egypt (see below). At least 170 million people are infected worldwide (Science 288: 339, 2000), at least 3 million in the USA, with about 10000 deaths yearly (that mortality figure is conservative and is going to increase: JAMA 297: 784, 2007).
Hepatitis C virus is transmitted by the same routes as hepatitis B, but is clearly not nearly so catching. The best route seems to be blood transfusion or needle-sharing (J. Inf. Dis. 162: 823, 1990; hemophiliacs Blood 84: 1020, 1994).
Needlesticks from infected blood carry about a 6% chance of infection (Br. Med. J. 315: 333, 1997) and prophylactic treatment with anti-hepatitis C medicines is now administered routinely after such events.
Strangely, nobody yet knows the prevalence of perinatally-transmitted hepatitis C, but it can declare itself later in life as a fulminant illness: Arch. Dis. Child. 88: 160, 2003.
Thankfully, with changing lifestyles (maybe) and surveillance in the hospital (certainly), the transmission rate of hepatitis C is only about 1/5 what it was in the 1980's (Sci. Am. 280(3): 17, March 1999.)
The risk from a transfusion is now about 1 in 2 million (Lancet 361: 161, 2003).
* Among several hundred Irish women infected in the '70's by bad RhoGam, half still had demonstrable virus, most of these had some inflammation, many had some fibrosis, but only two had cirrhosis (NEJM 340: 1228, 1999).
Hepatitis C transmission by acupuncture is now so well-known that you'd do well to warn your patients to be sure they know who's doing it (Can. Fam. Phys. 49: 985, 2003).
Sexual transmission is very rare but it probably occurs (JAMA 269: 361 and 392, 1993; Gut 45: 7, 1999; the risk to MSM's seems very low Am. J. Pub. Health 95: 502, 2005); older studies suggest that around a quarter of spouses do eventually catch it (Ann. Int. Med. 120: 748, 1994). Vertical transmission from Mom is common, especially if Mom has lots of virus on board: NEJM 330: 744, 1994. In some communities -- in striking contrast to HIV, hepatitis B, and so forth -- around 40% of people who carry the virus haven't got a clue how they got it.
In the US, if you're living clean enough to donate blood, your chance of coming down with hepatitis C is very low (BMJ 316: 1413, 1998; NEJM 341: 556, 1999).
A majority of people with the antibody do have detectable virus by PCR in the blood.
* Unlike hepatitis B and D, to date there is no reliable stain for hepatitis C.
The presence of the virus's RNA in the blood (or liver tissue, which is harder to obtain) is today's standard for proving infection. The exact specificity of a positive antibody depends on the test (the cheap first-generation assays were infamously nonspecific) and of course the population screened (clean-living people's positive screens are more likely to be false than needle-drug-user's positive screens). For an update on the difficult subject of who to screen and how, see Am. J. Gastro. 100: 607, 2005.
We used to teach that only a few folks do clear the virus quickly after being infected (Science 288: 333, 2000); with newer methods of detecting acute infection, some folks say that around 67% clear themselves without treatment within 8 weeks (Hepatology 37: 60, 2003), though it's also known that many people's viral levels simply become undetectable and the infection does recur. There's still a question of whether to treat immediately or wait-and-see. And some people who are chronically infected have viremia only intermittently, and in some studies, rates of spontaneous clearance over the years are relatively high (Irish J. Med. Sci. 174: 37, 2005 gives a surprising 31%, perhaps just the luck o' the Irish.)
* Children exposed from blood transfusion do much better, often clearing themselves (NEJM 341: 912, 1999).
Egypt, with around 20% of its people infected, has the highest rate.
Probably because of needles (used to administer antimony
in the treatment of schistosomiasis![]() )
not being sterilized between patients during one of the aggressive
"eradicate schistosomiasis" campaigns (Lancet 355: 887, 2000).
)
not being sterilized between patients during one of the aggressive
"eradicate schistosomiasis" campaigns (Lancet 355: 887, 2000).
Incubation period is a week to six months; texts give the average as 8 weeks. The acute infection is more likely to be subclinical (or cause minor "belly trouble"), and massive necrosis does not occur. However, infection usually becomes progressive. Nobody knows the rate of subclinical infections, so nobody truly knows how many people clear themselves of the infection in the acute phase. If you don't clear it, fortunately, progression is slow, and severe liver failure results in only about 10-30% of people and usually only after decades.
* Miraversin, an antisense oligonucleotide for hepatitis C (miravirsen): NEJM 368: 1685, 2013.
You can get sick several times if you get a big dose of the bug several times (Lancet 343: 388, 1994). After acquiring the virus via blood transfusion, chronic infection with abnormal liver histology happens more often than not (Ann. Int. Med. 137: 961, 2002). The impact on overall length of life is usually small (NEJM 327: 1906, 1992; Gut 47: 845, 2000) but the infection is still a serious business. Patients who are treated for their illness cost third-party payers much less than those who do not (lost productivity, advanced disease): Dig. Dis. Sci. 57: 2995, 2012.
Around a third of hepatitis C virus carriers have aggressive-looking chronic hepatitis or cirrhosis (Br. Med. J. 308: 695, 1994). Unlike the other viral diseases, there is often quite a bit of fatty change (correlates with severity: J. Inf. Dis. 192: 1943, 2005 -- some strains produce random fat-laden cells, others cause the metabolic syndrome itself with most cells in zone 3) and/or regenerative change in the bile ducts, rather few inflammatory cells (maybe just lymphoid aggregates) in the parenchyma, and a portal infiltrate that's all lymphocytes (no plasma cells or eosinophils, and perhaps even making a "nice lymphoid aggregate" or even a "nice lymphoid follicle / germinal center") suggests hepatitis C. The progression to fibrosis is steady and usually takes decades; if you're male, a drinker, older, and/or unlucky, it may take only a decade or so (Lancet 349: 825, 1997.)
NOTE: As with hepatitis B, carrying hepatitis C, with or without ongoing liver disease, is an important cause of cryoglobulinemia (Am. J. Med. 96: 124, 1994; NEJM 330: 751, 1994 for the success of alpha-IF therapy). The cryoglobulins are immune complexes made of the virus and the antibodies.
* How does hepatitis C virus produce fibrosis? There is often remarkably little inflammation. In one model, the virally-infected hepatocytes produce huge amounts of transforming growth factor beta, causing stellate cells to produce collagen. Stay tuned; this may become the basis for a novel anti-fibrogenic therapy (Gastroenterology 129: 246, 2005).
* NOTE: Hepatitis C virus tends to drive out hepatitis B virus over the long-term in patients infected with both (Gastroent. 106: 1048, 1994, others).
* As with hepatiis B, quite a few of these people never have elevated transaminases, even as they progress to cirrhosis. We have to wonder how these people's infections were detected (Am. J. Gastro. 98: 1588, 2003).
We now eliminate about half of chronic infections using a combination of pegylated interferon and ribavirin. This is one of the most important triumphs of late-20th_century medicine in the last decade. More about this on your gastroenterology rotation.
Ishak fibrosis score:
For the harder-to-treat HCV genotypes, a metavir score of F2 or less (few or no septa) or a Ishak score of 2 or less (no bridging) may justify following but not treating the patient.
Like hepatitis B, longstanding infection with hepatitis C places a person at grave risk for hepatocellular carcinoma (Lancet 345: 413, 1995).
Nowadays, hepatitis&C infections are treated aggressively with the hope of cure. The difficult genotype 1 seems to respond well to the regimen with added boceprevir (NEJM 364: 1195 & 1207, 2011 -- now there are reports of most patients having their infections cleared with ABT-450 -- protease inhibitor -- plus retonavir NEJM 370: 222, 2014). For people with genotypes 1, 2 or 3 even with previous treatment failures, the combination of daclatasvir and sofobuvir seems to clear most infections NEJM 370: 211, 2014. Ledipasvir and sofosbuvir: NEJM 370: 1483, 1879 & 1889, 2014. Ombitasvir, dasabuvir and ribavirin: NEJM 270: 1594 and 1604, 2014. Oral regimen: Daclatasvir, asunaprevir, and beclabuvir: JAMA 313: 1728, 2015. More about the emerging regimens, which can work even for the hard-to-treat genetype 1 strain: Lancet 385: 1107, 2015. You can read about a variety of other current studies.
Immunology:
In contrast to hepatitis B, the presence of ANTI-HCV usually indicates the persistent presence of hepatitis C virus in the body. The original work found that around 60% of people found to be positive with the first-generation antibody assay did indeed have virus detectable by PCR. The newer assays (RIBA) are much more specific, and are used if the screening is positive but no virus is present in the blood -- if these are positive, the person has probably self-cured.
* There's hope that we'll have a hepatitis C vaccine, but it's a long way off. Like HIV, the virus is notorious for mutating rapidly, even in the same patient, and this isn't good for vaccine-makers. And like HIV, antibodies aren't very protective. Updates J. Imm. 176: 6065, 2006; Gastroenterology 130: 453, 2006.
|
* Echazabal vs. Chevron. Mario Echazabal had hepatitis C, and Chevron
refused to allow him to work around chemicals that might be hepatotoxic.
He claimed that this violated his rights under the Americans with Disabilities
Act. His supporters accused Chevron of "paternalism", which as you know
is currently anathema in many circles but is (like it or not) the foundation
of occupational health and safety policy.
The Supreme Court decided unanimously for
Chevron in 2002. I agree.
See Am. J. Pub. Health. 93: 540, 2003. |
HEPATITIS G and the HEPATITIS GC family are hepatitis-C-like flaviviruses.
Hepatitis G virus and its cousin GB virus C are relatively common infectious agents that produces a chronic viremia. They're now considered two isolates of the same virus "GB Virus C". They are known to be transmitted by blood products, sex, needles, and mother-to-child (Arch. Dis. Child. 80: F72, 1999). There's an antibody test (Lancet 349: 318, 1997).
Whether these critters ever make anybody sick has been under study for two decades There doesn't seem to be an acute illness (NEJM 336: 741 & 747, 1997). More studies failing to show any evidence that they make you sick: Gut 103: 103, 1998; Arch. Dis. Child. 80: F72, 1999; Ann. Int. Med. 126: 874, 1997.
A person may clear the virus, or have persistent virus infection. Around 85% of hepatitis C patients have evidence of past or present infection (J. Inf. Dis. 194: 410, 2006).
The virus also multiplies in B- and T-lymphocytes (J. Inf. Dis. 193: 451, 2006). It inhibits HIV replication in vitro (J. Inf. Dis. 192: 2147, 2005; Lancet 363: 2040, 2004), and it is claimed that persistent GB virus C coinfection slows the rate of progression of HIV disease (J. Inf. Dis. 194: 410, 2006; NEJM 350: 981, 2004), Stay tuned.
* People who study these things say that C, G, and the GC's all evolved from yellow fever or dengue fairly recently.
* TTV ("transfusion transmitted virus") is a DNA virus that's very common (10% of folks) in Japan, less common in the West. It elevates transaminases after a transfusion, but nobody's found anyone sick from it yet (Lancet 352: 164, 1998).
HEPATITIS E: An important, epidemic calicivirus (now hepevirus) infection in the poor nations (Lancet 379: 2477, 2012; NEJM 367: 1237, 2012).
* This disease produced at least one major outbreak (New Delhi monsoon floods, 1955) but was officially discovered when it caused an outbreak at a Soviet military camp in Afghanistan in 1983. Dr. Mikhail S. Balayan, who knew he was immune to hepatitis A, mixed "a pooled faecal extract from affected soldiers" with his yogurt, went back to Moscow, and when he got sick found the new virus (named HEV) in his stool by electron microscopy Dr. Balayan won the Order of Lenin.
It's usually mild, but there are around 70,000 deaths worldwide each year from massive hepatic necrosis. For some reason, pregnant women (or those taking oral contraceptives: Am. J. Trop. Med. 82: 12, 2010) are likely to be severely affected, and may die. It never becomes chronic in healthy folks. There is no specific treatment. In outbreaks, mortality can be as high as 25%.
In the poor nations, it's waterborne. The developed world has different strains and probably these are from contaminated, poorly-cooked meat. In 2009, the fact that this infection is fairly common in France was linked to the practice of eating raw pig livers. I am not making this up (Emerg. Inf. Dis. 15: 110, 2009).
You'll make the diagnosis on the presence of IgM antibodies. Around 25% of people from the Middle East have had it, but it is less prevalent in the rest of the world (J. Inf. Dis. 16: 801, 1994). Review from the CDC in Inf. Dis. Clin. N.A. 14: 669, 2000.
The first cases of chronic hepatitis E, progressing to cirrhosis, were reported in transplant recipients in 2008 (NEJM 358: 811 & 859, 2008).
Vaccine NEJM 356: 895, 2007; Lancet 376: 849, 2010; NEJM 372: 899 & 914, 2015.
YELLOW FEVER: The reservoir is monkeys (South America and Africa) and it is transmitted monkey-to-human or human-to-human by the Aedes mosquito. The diagnosis is clinical. A relatively mild febrile illness with viremia lasts 3-6 days; in a minority, a fulminant hepatitis follows after initial remission. Pathologists see Councilman bodies, necrosis especially in the mid-zone of the lobule, and a surprising lack of inflammatory response (still the pattern: Trans. Royal. Soc. Trop. Med. 101: 831, 2007). Yellow fever today in Bolivia: Lancet 353: 1558, 1999. Death from yellow fever is probably not so much due to liver failure as to overactivation of cytokines, much as in sepsis: J. Inf. Dis. 190: 1821, 2004. The live yellow-fever vaccine is one of the oldest and most effective but kills some of the recipients; a new inactivated vaccine shows promise (NEJM 364: 1326, 2011). Yellow fever outbreak in the Darfur region of the Sudan: NEJM 368: 689, 2013.
![]() Yellow fever
Yellow fever
Yutaka Tsutsumi MD
CHRONIC HEPATITIS NOT CAUSED BY VIRUSES
AUTOIMMUNE ("lupoid"; "plasmacytic") HEPATITIS: Chronic hepatitis progressing to cirrhosis, without chronic virus infection but with evidence of immune injury. Reviews NEJM 354: 54, 2006; Lancet 382: 1433, 2013.
Poorly understood, but fairly common (prevalence of around 16 people in 100,000, much higher in some groups especially the Eskimo / Inuit), and deadly if missed (3/4 dead in ten years if untreated). We'll distinguish the different types (which bear little relationship to real lupus) when we discuss liver function testing. The most common type ("type I") features autoantibodies against smooth muscle / filamentous actin / F-actin. Other types are distinguished by other antibodies (anti-LKM-1 for the much more severe type II; anti-SLA/soluble liver antigen/LP/liver-pancreas once defined "type III" but now are a variant of type I: Gastroent. 135: 2107, 2008), but the havoc is wrought by the T-cells.
* Despite its reputation as a slow, chronic illness, it may be acute and even fulminant (Dig. Dis. Sci. 58: 897, 2013).
* Leave the diagnosis of the uncommon "autoantibody-negative autoimmune hepatitis" to the gastroenterologists. There may be an unknown autoantigen, or the familiar antibodies may show up later (Dig. Dis. Sci. 57: 610, 2012).
Current thinking is that something first damages the liver (probably one of the viral hepatitis family, or some drug or poison, or whatever), and patients then get sensitized to their livers and start destroying them over the long haul. More about this later.
Drugs that trigger "lupoid hepatitis" include some of the older ones, and today minocycline (Br. Med. J. 312: 169, 1996) and the infamous herbal remedy "dai-taiko-so".
Future pathologists: Autoimmune chronic hepatitis usually features a lot more plasma cells than does viral chronic hepatitis.
* F-actin antibodies plus a histologic picture of autoimmune plasmacytic hepatitis is fairly common in chronic hepatitis C infection and HCV/HIV coinfection (Am. J. Clin. Path. 314: 228, 2012). If you are serious about this being autoimmune hepatitis, ask for the ELISA (very sensitive, now will perhaps define the disease) rather than immunofluorescence.
Unlike in viral infection, the response to immunosuppression (i.e., glucocorticoids and for the long haul azathioprine) is generally good and the illness generally remits after a few years. (Anti-LMK-1 disease is less likely to respond and often does not remit and often relapses, requiring long-term maintenance). The common protocol, which often cures, is based on azathioprine (NEJM 333: 958 & 1004, 1995; update on why some patients tolerate it poorly: Dig. Dis. Sci. 51: 968, 2006).
We suggest following the course of the disease with biopsy, since liver enzymes correlate rather poorly with histologic progression.
* Future pathologists! Here's your scoring system (J. Hep. 31: 929, 1999)
About 85% of these people can achieve remission with today's treatments. Transplantation may eventually be necessary. After ten years, the transplant has about a 50/50 chance of being involved by recurrent disease (Gut 52: 893, 2003; update on the natural history of recurrent autoimmune hepatitis Dig. Dis. Sci. 57: 2248, 2012).
PRIMARY BILIARY CIRRHOSIS: An autoimmune disease caused (we don't know exactly how) by antibodies against pyruvate dehydrogenase ("anti-mitochondrial antibodies"). Lancet 362: 53, 2003. We'll talk more about this under "Liver Testing".
The bile ducts are selectively attacked by the immune system, eventually resulting in severe obstructive jaundice.
For some reason, the biliary epithelial cells express pyruvate dehydrogenase, or something very much like it, on their luminal surfaces (J. Clin. Invest. 91: 2653, 1993). This is evidently the target.
* The anti-nuclear antibodies often seen in primary biliary cirrhosis are usually directed at glycoprotein 210 and/or nucleoporin 62, on the nuclear surface pores.
* There are several known associations with HLA and Interleukin 12 genes: NEJM 360: 2544, 2009.
The histopathology begins with chronic inflammation (mostly portal, sometimes some interface hepatitis), and progresses through bile-duct obliteration and collateral formation to micronodular (why?) cirrhosis. For the details see Mayo Clin. Proc. 73: 179, 1998.
Less easy to explain are the frequent appearance of granulomas and Mallory's hyaline.
* Patients typically complain of severe fatigue, even early in the disease. One group attributes this to retained manganese (Gut 53: 587, 2004).
* Pitfall: Sarcoidosis can look exactly like PBC-with-granulomas, but the AMA is negative.
* Copper is likely to accumulate. Don't confuse it with Wilson's.
{24568} primary biliary cirrhosis, early; cirrhosis has not really developed yet, but portal areas are
inflamed; you could not tell at this magnification that this is primary biliary cirrhosis
{24569} primary biliary cirrhosis, histology (i.e., the bile duct is gone)
|
|
|
|
|
Primary biliary cirrhosis was found in the 1990's to be considerably more common than we had once thought, and to responds to therapy with bile salt analogues such as ursodeoxycholic acid (nobody knows why; Br. Med. J. 312: 1181, 1996) -- most cases get fairly good response; if more is needed, methotrexate and colchicine are helpful (Dig. Dis. Sci. 55: 3291, 2010).
* "Primary autoimmune cholangitis" looks something like primary biliary cirrhosis, but has high ANA titers and no anti-mitochondrial antibodies. See Am. J. Surg. Path. 18: 91, 1994; update on sorting out the autoimmune hepatitis family histologically Am. J. Clin. Path. 114: 705, 2000.
* IDIOPATHIC ADULTHOOD DUCTOPENIA is disappearance of the interlobular bile ducts; it may be asymptomatic (elevated GGT prompts its discovery) or progress to cirrhosis (NEJM 336: 835, 1997). More often, this is the result of drug-induced cholestasis that isn't recognized in time.
"Secondary biliary cirrhosis" is more likely to be just fibrosis, due to obstruction of the common bile duct, usually in chronic pancreatitis. If the stenosis is relieved, the fibrosis often regresses some (NEJM 344: 452, 2001).
DRUGS AND POISONS (a problem easily overlooked; reviews Gut 44: 731, 1999, NEJM 354: 731, 2006; including how to establish the relationship and warnings about what will happen if you leave the patient on the medication that's causing the liver disease)
As before, "all poisons are drugs, all drugs are poisons".
To make the call, you want to see one of these:
Especially rough on the liver:
 |
Any of these can produce massive hepatic necrosis. The toadstool and the acetaminophen overdose will produce massive hepatic necrosis; "Ecstasy" in recreational amounts is famous for the same (Transplant. Proc. 33: 2743, 2001). The others are more likely to produce a hepatitis-like picture.
ACETAMINOPHEN OVERDOSE is very common.
The drug is metabolized by two different pathways, one "safe", the other productive of noxious free radicals. Ordinarily, we use only the "safe" pathway, but when that is overloaded, the drug gets shunted into the bad pathway.
* We've already seen the "two-pathway" concept in our discussions of atherosclerosis and Alzheimer's disease. Stay tuned for the discovery of more "two-pathway diseases".
Three or four days after the overdose, the patient gets sick and lapses into hepatic failure. By this time, the drug itself may be mostly gone.
Future emergency room specialists: You can block the "bad" pathway using good old N-acetyl-cysteine, or "Mucomist", from the respiratory care department.
On biopsy or at autopsy, the necrosis is most obvious in the central areas. There's little or no inflammation.
* In England and Wales, acetaminophen is the popular method of suicide. Clever legislators required selling acetaminophen ("paracetamol") in smaller packages that required folks to unwrap each tablet. The suicide rate dropped dramatically (BMJ 346: f403, 2013).
CHOLANGITIS
BACTERIAL CHOLANGITIS ("ascending cholangitis", etc.) is suppuration involving bile ducts.
![]() Ascending cholangitis / biliary obstruction
Ascending cholangitis / biliary obstruction
Joel K. Greenson MD
U. of Michigan
The underlying cause is almost always obstruction. Pretty much any gut bacterium can be the opportunist. E. coli is most common; clostridial gas gangrene of the liver is ultra-deadly.
As you would expect, patients are super-sick with the acute infection. Call a surgeon, since the bile has to be drained.
The give-away on histologic exam is neutrophils within the bile ducts. Since there's obstruction, look for bile plugs, too.
LIVER ABSCESSES, in the U.S. are usually of bacterial origin, spreading either up the bile ducts ("cholangitis abscess" -- ascending cholangitis) or via the portal vein ("pyelophlebitic abscess" -- appendicitis, diverticulitis), or from septic emboli (bacterial endocarditis), or following a dirty wound.
Naturally, patients are super-sick, as with ascending cholangitis.
"Amebic abscesses", a misnomer, are areas of "anchovy paste" necrosis without much inflammation. Hydatid cysts can become infected, forming real abscesses.
|
|
* PERICHOLANGITIS has been discarded as a term in pathology. Inflammation AROUND the bile ducts, typically extra lymphocytes, is very common at autopsy and no one knows what it means, if anything.
One could conjecture that the liver clears the blood of foul products of fatal disease, and that these are excreted in the bile and attract inflammatory cells.
Patients with ulcerative colitis and Crohn's generally have fibrosis around the bile ducts (SCLEROSING CHOLANGITIS) and often biliary obstruction / biliary cirrhosis.
Or one can have idiopathic PRIMARY SCLEROSING CHOLANGITIS, a curious, probably-immune-mediated entity. The extra-hepatic bile ducts in these diseases come to look like uneven strings of beads. Under the microscope, there is onionskin fibrosis around the intrahepatic bile ducts. Review Lancet 382: 1587, 2013.
More recently, there are claims that there are distinctive autoantibodies against biliary epithelium (Gastroent. 132: 1504, 2007), and this seems to be holding up.
Medical therapy does not slow the progression of this disease. Many of these people need liver transplants, and the disease tends to recur in the transplanted organ.
* Sclerosis of the bile ducts is one feature of IgG4 sclerosing disease. More about this when we understand it.
* Perhaps 20% of folks go on to develop cholangiocarcinoma.
|
|
|
OTHER INFECTIONS:
KALA-AZAR (a vicious form of leishmaniasis![]() )
packs Kupffer cells with organisms but does not interfere with liver function.
)
packs Kupffer cells with organisms but does not interfere with liver function.
INFECTIOUS MONO from any of the usual causes can produce elevated transaminases and/or mild hepatocyte failure.
SECONDARY SYPHILIS![]() can give an acute hepatitis,
while TERTIARY and especially CONGENITAL syphilis are noted causes of hepar lobatum, due to scar contraction.
can give an acute hepatitis,
while TERTIARY and especially CONGENITAL syphilis are noted causes of hepar lobatum, due to scar contraction.
Don't forget LEPTOSPIRA in unexplained jaundice, especially if there is hemolysis.
* PENICILLIUM MARNEFFEI is an opportunist in AIDS, especially in Southeast Asia (Arch. Path. Lab. Med. 128: 191, 2004).
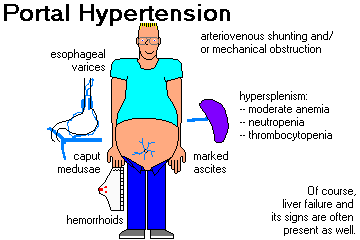
Increased pressure in the portal venous system, for whatever reason (usually increased resistance to flow and/or increased anastomoses with the arterial circulation).
PRE-HEPATIC CAUSES
Portal vein obstruction / compression
Thrombus (typically from tumor invasion, hypercoagulability, or polycythemia vera)
Tumors (hepatic, biliary, pancreatic)
Really bad pancreatitis
INTRA-HEPATIC CAUSES
Cirrhosis (both fibrosis of the vasculature, especially hepatic veins, and AV-shunting contribute)
* Central hyaline sclerosis without cirrhosis
Really bad fatty change
Alcoholism / "alcoholic hepatitis", etc.
NASH
Reye's
Schistosomiasis![]() (eggs plug portal vein radicles, leading to the infamous "pipestem fibrosis")
(eggs plug portal vein radicles, leading to the infamous "pipestem fibrosis")
* Sarcoid / TB![]()
* Congenital hepatic fibrosis (a thankfully-rare birth defect, with very few veins in the expanded portal areas; one known locus is a variant autosomal recessive polycystic kidney disease gene, and hepatic fibrosis is common in the fully-expressed condition as well: Medicine 85: 1, 2006)
Osler-Weber-Rendu telangiectasisa (abnormal vascular communications: NEJM 343: 931, 2000).
"Idiopathic" (dubious)
POST-HEPATIC CAUSES
Budd-Chiari
Constrictive pericarditis
Tricuspid insufficiency
Really bad right-sided heart failure
Regardless of cause, portal hypertension is troublesome.
Patients get ASCITES, or large accumulations of fluid in the abdomen. This is troublesome. Mechanisms of formation include
(1) the obvious increase in hydrostatic pressure in the venules;
(2) the increase (most mechanisms) in hydrostatic pressure within the hepatic sinusoids themselves (the "increased hepatic lymph formation" of "Big Robbins"; this stuff is likely to be rich in protein, since the hepatic sinusoidal "endothelium" is discontinuous)
(3) diminished circulatory volume due to low serum albumin, with a tendency of the kidneys to retain sodium and water. (BEWARE! If you give these patients a diuretic, you can send them into shock, kidney failure, "hepatorenal syndrome", etc., etc.)
{19381} ascites
{05952} ascites in a known cirrhotic
{05953} ascites in a known cirrhotic
|
|
PORTO-SYSTEMIC SHUNTING results when blood from the guts finds other routes back to the right side of the heart.
This results in ESOPHAGEAL VARICES (which can bleed profusely), SEVERE HEMORRHOIDS ("anorectal varices", which can bleed profusely), and the distinctive CAPUT MEDUSAE around the belly-button.
* This also probably is the cause of the usually-mild IMMUNOGLOBULIN A NEPHROPATHY typical of cirrhotics (i.e., asymptomatic hematuria). IgA from the gut ends up in the kidneys, rather than being cleared by the liver.
FIBROCONGESTIVE SPLENOMEGALY produces big, firm spleens that often produce clinically significant hypersplenism (i.e., they make the person anemic, neutropenic, and/or thrombocytopenic). This is bad.
You can treat portal hypertension effectively by doing a porto-caval shunt operation. If the underlying problem is cirrhosis, this will result in blood flowing directly from the bowel to the systemic circulation, making hepatic encephalopathy much, much worse ("portosystemic encephalopathy"). But most people prefer this to dying of bleeding varices.
Sclerosing agents save the lives of patients during acute bleeds. Today, banding ("band ligation") is doing the same (Br. J. Surg. 86: 437, 1999.)
For lasting control, the patient is likely to have the shunt placed inside the liver itself, by the radiologist who passes a catheter down the jugular vein ("transjugular intrahepatic portosystemic shunt" -- now revolutionized by the development of the covered shunt which allows longer shunt patency AJR 199: 746, 2012).
* Old-fashioned "prophylactic sclerotherapy" of esophageal varices actually increased the risk of dying. Perhaps it just made whatever vein didn't get sclerosed into a bigger varix. NEJM 324: 1779, 1991.
Of course, portal hypertension isn't the only problem that the cirrhotic has. See "When the liver fails", above.
ALCOHOLIC LIVER DISEASE (Lancet 345: 227, 1995; Mayo Clin. Proc. 76:
1021, 2001) 
|
Woe to those who demand strong drink as soon as they rise in the morning, and linger into the night while wine inflames them!
-- Isaiah 5:11
Sir, I have known more old drunkards than old doctors.
-- Dr. Rabelais

Everybody knows alcohol is bad for the liver, but there is perennial confusion about the various patterns of liver injury and their outcome.
ALCOHOLIC STEATOSIS ("alcoholic fatty liver")
We've already reviewed why fat accumulates in liver cells damaged by alcohol.
To review: The drunken hepatocytes make too much fatty acid, make it into excess triglyceride instead of burning it, then can't complex the triglyceride to apolipoproteins, and can't export the lipoproteins they do make.
Current thinking supports popular wisdom that alcohol itself does the damage (in fatty change and the more severe forms of "alcoholic liver disease"). Poor nutrition doesn't help, either (i.e., few people make daily trips to the salad bar while on benders.)
If you've ever drunk a case of beer over a great football weekend, I bet you've had fatty liver. Did you notice? Probably not. The "disease" is usually just a pathology finding, unless:
Fatty liver is, by itself, completely reversible once the drinker sobers up. The same applies to fatty liver from other causes i.e., after bariatric surgery (Gastroenterology 130: 1617, 2006), in obesity, in ill-controlled adult-onset diabetes, in problem pregnancy, in galactosemia, in methotrexate toxicity, in Wilson's disease, and as a complication of a few rare disorders of lipid metabolism.
{49271} fatty cirrhotic liver vs. normal
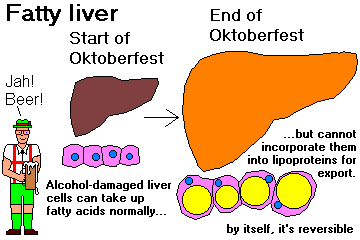
NOTE: "Microvesicular steatosis" (smaller vacuoles, usually several per cell) is typical of Reye's syndrome, problem pregnancies, mitochondrial problems (Gastroent. 108: 193, 1995), and toxicity from outdated-tetracycline poisoning, or some of the HAART drugs. It's also reversible; really, "microvesicular" steatosis can occur in any fatty liver.
* In my opinion, the "microvesicular vs. macrovesicular" distinction is of no value. I have seen only one Reye's autopsy, and it was pure macrovesicular fat. I've autopsied many, many problem drinkers, and the pattern is often mixed, and sometimes microvesicular-only. I addressed this question to the pathology on-line group in 1996, and there was no disagreement from a few hundred experienced pathologists.
NOTE: "Fatty change" confined to the stellate cells ("Ito cells") is vitamin A overdose. "Stellate cell lipidosis" is often due to vitamin A overdose or Retin-A: Am. J. Clin. Path. 119: 254, 2003.
* Of course, if you continue drinking, you're at serious risk to get cirrhosis, even if "you only had fatty change" on a biopsy. Bad prognostic indicators include giant mitochondria and mixed microvesicular and macrovesicular fat. See Lancet 346: 987, 1995.
ALCOHOLIC HEPATITIS (NEJM 360: 2758, 2009)
Much more serious. Here, we have inflamed liver cells with widespread liver cell death and some stranger things than fat in the liver cells. The process is worst in the centrilobular regions.
Alcoholic hepatitis comes on suddenly, usually after years of problem drinking. However, it's clearly a different entity from both common "fatty liver" and the all-too-familiar cirrhosis. The patient -- who may not even have had a drink for a few weeks -- suddenly comes in with jaundice (bilirubin 5 mg/dL or more), elevated transaminases, and signs of portal hypertension and liver failure. Typically there's also an increase in blood neutrophils.
The cause of alcoholic hepatitis -- the sudden appearance of a new disease after years of drinking -- has been a minor mystery of medicine for centuries. Today's thinking focuses on alteration of the permeability of the large bowel, allowing endotoxin to flow into the portal circulation. It activates Kupffer cells and causes them to produce TNF-α.
The process can kill a patient directly. If an anesthetic is required, this may precipitate liver failure. Or some Hippocrates who doesn't want to biposy the liver because of the known tendency to bleed will feel it to be enlarged and assign an erroneous diagnosis of cirrhosis.
On biopsy, surviving liver cells often bear MALLORY'S ALCOHOLIC HYALINE (not pathognomonic, but suggestive). This is masses of altered prekeratin fibers plus stress proteins. Free Mallory's hyaline is chemotactic for neutrophils.
Also look for GIANT MITOCHONDRIA, (* Yokoo bodies, after one of the professors who trained your lecturer), PAS-negative blobs in the cytoplasm.
* Mitochondria (giant or no) in alcoholics' livers often have genetic damage, presumably because alcohol generates toxic free radicals inside them (Gastroent. 108: 193, 1995). This is one more putative mechanism of fatty liver. How?
The process tends to be worse in the central regions, but no area is spared. Cholestasis is usual because of compromise of bile canaliculi, with bile lakes and bile plugs. At the same time, the bile ducts may proliferate within the portal areas.
As the liver cells die off, look for FIBROSIS, notably around the central veins ("central hyaline sclerosis"; see Virch. Archiv. 614: 11, 1989; Postgrad. Med. J. 76: 280, 2000). This won't go away, and seems to be a rapid route to cirrhosis. (* You may be told that alcoholic hepatitis always precedes alcoholic cirrhosis; there's no reason to think this is true.)
{39929} alcoholic hepatitis, good Mallory's hyaline
{26702} alcoholic hepatitis, good Mallory's hyaline
{08832} alcoholic hepatitis with fibrosis; there is no
good hyaline; cells are becoming entrapped in the fibrous tissue
{21056} "alcoholic hepatitis", looks like early cirrhosis to me
{26693} "alcoholic hepatitis", looks like early cirrhosis to me
![]() Alcoholic hepatitis
Alcoholic hepatitis
Alabama
Wikimedia Commons
The treatment is sobriety, and perhaps medications or lactobacilli to alter the gut flora; watch for TNF-blockers.
Again, the histology is not pathognomonic for alcoholism. The heart-drug amiodarone in particular is infamous for producing the same histopathology, and post-ileal bypass hepatitis, Wilson's disease (shouldn't miss this one!), NASH, and * East Indian childhood cirrhosis (copper toxicity in the genetically-predisposed; J. Path. 195: 264, 2001) can be dead-ringers.
If you are actually in the hospital and your primary diagnosis is alcoholic hepatitis, you probably have it very, very bad... explaining why the mortality figures in some studies are so high (for example, J. Clin. Gastro. 40: 833, 2006).
Fortunately, if you sober up, all that will remain is whatever minor scarring has occurred. The liver cells will regenerate nicely, and your liver will probably be fine.
ALCOHOLIC CIRRHOSIS ("Laennec's cirrhosis", other names)
Exactly what causes the progression (if it is a progression) from reversible changes (fatty change, Mallory bodies, inflammation) to irreversible (?) disease (i.e., fibrosis-cirrhosis) is obscure.
Easy to remember -- if you drink less than six beers per day / four glasses of wine per day, cirrhosis is unlikely. In order to get cirrhosis, one needs at least 15 pint-years (i.e, a pint of the hard stuff per day for fifteen years, three pints a day for five years, or similar). Many cirrhotics have much more. Yet 2/3 of heavy drinkers die without cirrhosis. Nobody knows why.
At first, the liver is big because of widespread hepatocyte overgrowth and fatty change from ongoing drinking. Later, with advanced scarring and enforced sobriety, the liver becomes rather small.
Microscopically, you'll see fibrosis and nodules instead of the normal architecture and proliferated bile ducts in the scar tissue (a good sign of alcoholism).
Early Laennec's cirrhosis has fine bands and a micronodular pattern. Late, the pattern becomes post-necrotic. When the scar tissue starts seriously obstructing bile flow, clinicians see jaundice and the pathologist sees bile plugs.
{10538} alcoholic cirrhosis, gross
{10844} alcoholic cirrhosis, gross
{18795} alcoholic cirrhosis, gross
{20163} alcoholic cirrhosis, histology; trichrome stain
{08835} alcoholic cirrhosis
{40412} alcoholic cirrhosis
{10535} "alcoholic cirrhosis" closeup
of a lobule; note the Mallory's hyaline and the neutrophils
attacking it. There may be fibrosis
elsewhere, but this merely looks like alcoholic hepatitis.
{39593} alcoholic cirrhosis; lots of bile
duct proliferation (as is usual in alcoholic cirrhosis);
edge of a nodule on each side of the screen
The cirrhotic must decide what he or she wants out of life. Continuing drinking is overall as lethal as untreated AIDS. Sobriety gives maybe a 90% chance of not dying of the cirrhosis within the next five years.
|  |
NON-ALCOHOLIC FATTY LIVER DISEASE -- non-alcoholic steatohepatitis, NASH, non-alcoholic fatty liver diseae, NAFLD (Am. Fam. Phys. 73: 1961, 2006; pathologists see Am. J. Clin. Path. 128: 837, 2007; J. Clin. Path. 60: 1384, 2007; Gastroent. 134: 1682, 2008; NEJM 363: 1341, 2010); BMJ 343: d3897, 2011; Gut 60: 1152, 2011 -- "what's new under the microscope"); JAMA 313: 2263, 2015.
NASH -- non-alcoholic steatohepatitis, fatty change into which acute inflammation, necrosis and fibrosis can creep -- is a poorly-understood but very real, very common condition that is only now getting the attention it deserves.
In countries where little alcohol is consumed and there
is not much hepatitis or schistosomiasis![]() ,
this is the most prevalent liver disease.
,
this is the most prevalent liver disease.
The anatomic pathology is much like alcoholic liver disease, with fatty change, often Mallory hyaline, and sometimes even cirrhosis (not so vicious as other forms of cirrhosis, but still sometimes requiring transplant -- Hepatology 43: 682, 2006).
There is always insulin resistance as well. Presently, discussions of pathophysiology emphasize the "adipokines" TNF-α and leptin, produced by bodyfat and causing insulin resistance. TNF-α is considered responsible for the cell injury, and leptin for fibrosis. The whole business is still very puzzling (Am. J. Path. 169: 846, 2006; Dig. Dis. Sci. 53: 1099, 2008); new players Gastroent. 134: 556, 2008; Am. J. Clin. Nutr. 89: 558, 2009.
The disease now easily the most common chronic liver disease in children and teens in the industrialized world, and many of these kids have fibrosis (Gastroent. 135: 1961, e2, 2008; Gastroent. 136: 160, 2009).
The mainstay of therapy has historically been weight loss and exercise
in the overweight, masterful inactivity and perhaps metformin in the non-overweight
(Med. Clin. N.A. 80: 1147, 1996; rationale for metformin Am. J. Gastro. 98: 2093, 2003).
Exercise works for the mouse model (Diabetes 60: 2720, 2011).
Betaine as a remedy: Am. J. Gastro. 96:
2534, 2001; Am. J. Gastro. The animal model (Am. J. Clin. Nutr. 79: 502, 2004) is
straightforward -- give rats a very high-lipid diet, let them eat as much
as they want, and they get NASH, with steatosis, necrosis, inflammation,
abnormal mitochondria (lost cristae, rarified matrix), and greatly elevated TNF-alpha production in the liver.
* CK18 ("apoptotic caspase"), the portion of cytokeratin cleaved by caspase 3 during
apoptosis, is a marker for both generalized total-body apoptosis and particularly
steatohepatitis (as opposed to just fatty liver in fat people -- J. Clin. Endo. Metab. 95: 343, 2010).
As you know well, not everybody tells the truth about their alcohol intake.
Mayo's has a formula using other labs that distinguishes boozers from
NASH patients (Gastroent. 131: 1057, 2006). High MCV (mean corpuscular
volume), and high AST/ALT ratio
mean alcoholism; staining of the biopsy specimen for PTP1b (protein
tyrosine phosphatase 1b) means NASH (see also Am. J.
Clin. Path. 123: 503, 2005). Cholestasis on biopsy is familiar
from alcoholic hepatitis, but is supposedly not seen in NASH (NEJM 360:
2758, 2009). If you have a fat, non-exercising heavy drinker, you may have both.
And so forth.
* Supposedly 30% of Americans have at least fatty liver from overweight
and inactivity ("mild NASH"). Fortunately or unfortunately, you
will diagnose this on scan or enzymes ("transaminasitis" after you've
ruled out
alcohol abuse, drug allergy,
hepatitis B, hepatitis C, autoimmune hepatitis,
antitrypsin deficiency, hemochromatosis, and Wilson's.) That is a HUGE
number of patients, and the burning question is, "Who do I send to a liver
specialist so that they don't get fibrosis and die / sue me?"
The mathematics people have come to the rescue. There are a group of
indices that crunch
lab values -- including AST (SGOT), ALT (SGPT), platelet count,
serum albumin fasting glucose, waistline/"body mass index", serum hyaluronic acid, serum tissue-inhibitor-of-matrix-metalloproteinases-1, serum aminoterminal propeptide of procollagen bype III,
and age. These indices will tell you who you do not need to refer to a liver
specialist (Gut 59: 1245 & 1265, 2010). By the way, what's the liver specialist going to do?
Get the patient to diet and exercise? Good luck. Prescribe metformin and
vitamin E? You the primary care physician could have done that. This will get stranger and stranger.
Before you lose sleep over the fact that it may not be easy to tell alcoholic
hepatitis from NASH, or who's going to get really sick from either,
please reflect that the mainstay of treatment for both is a healthy lifestyle.
So to a first approximation, "the treatment of both is the same."
* "Not to be confused with NASH" is the rare "glycogenic hepatopathy",
a big liver and elevated liver enzymes in a noncompliant type I diabetic.
The cells are overloaded with glycogen. Hopefully you'll never see it.
* See
also Gastroenterologist 5: 316, 1997; Gastroenterology 120:
1183 & 1281, 2001; the latter shows crystalloids in mitochondria
from these people that are not present in most controls, and suggests
that this is a mitochondrial disease that expresses itself in the
setting of obesity. (More on adult-onset diabetes itself as a likely
mitochondriopathy later.) We await confirmation.
Big review emphasizing how little is really known: Gastroenterology 122: 1649, 2002.
NASH as a cause of what used to be called "cryptogenic" cirrhosis: JAMA 289: 3000, 2003; 1/3 of patients will
get fibrosis, and 1/9 will get rapid progression to cirrhosis Am. J. Gastro. 98: 2042, 2003.
NASH is rampant among America's fat children (Am. J. Clin. Path. 127:
954, 2007), Korea's fat children (Dis. Dis. Sci. 54: 2225, 2009),
Italy's fat children (Trans. Res. 156: 229, 2010),
and we are now recognizing it as a fairly common cause of
death among young adults (Am. J. Clin. Path. 127: 20, 2007).
IRON OVERLOAD (update NEJM 350: 2383, 2004; Gastroenterology 139: 393, 2010; NEJM 366: 348, 2012; Dig. Dis. Sci. 57: 2988, 2012)
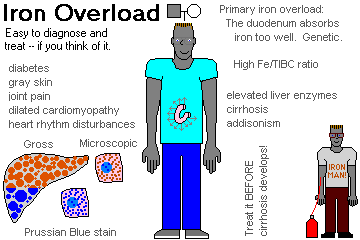
{49253} hemochromatosis causing cirrhosis (Prussian Blue stain, of course)
The human body cannot make iron atoms (see Science 226: 922, 1984 for how Mother Nature does it). We must get what we need from outside. The healthy adult has total body iron content of around 4 gm. The red cells contain close to 3 gm total. The cytochromes and other redox enzymes together contain much less than a gram, and most people have some stored iron. An iron storage problem is definitely present when the amount of iron in the body exceeds 10 gm.
A reasonably normal diet contains at least 10 mg of iron daily. This is more than adequate to replace a healthy man's daily losses, and is enough for a non-pregnant woman who does not have heavy menstrual flow. Heme iron is absorbed much more readily than non-heme iron, especially non-heme iron that is complexed with certain small organic molecules. (This explains why what worked for Popeye doesn't work for us.) Dietary iron deficiency anemia can be expected when a girl has been menstruating for about a year while consuming mostly twinkies, french fries, and diet Pepsi. Pregnant women, zealous blood donors, people with diseases in which blood loss (GI, urinary, uterine) cannot be easily controlled, and some growing children are the only people who might need to take iron supplements. (In normal adults, "prophylactic" iron supplementation can only mask serious disease. Taking iron supplements when they are not indicated kills people by delaying the diagnosis of GI and gynecologic cancers. One iron supplement in particular was famous for decades for its TV ads warning of "iron-poor tired blood". Fortunately for the public, the heavily-advertised nostrum used ferric iron, which makes the stool black so people would know the medicine was working, didn't upset the tummy very much, and wasn't absorbed very much (there's a duodenal mucosal cytochrome that's required to reduce ferric to ferrous iron to promote absorption). That's nice.) Healthy people absorb and lose around 1.0 mg of iron daily (1.5 mg for menstruating women).
Ferrous (Fe+2), and not ferric (Fe+3) iron, is absorbed across the "mucosal barrier", mostly in the duodenum. It is complexed to a protein called NRAMP-2 (no diseases here yet) that transports much more efficiently when body iron stores are low. So the amount of iron absorbed varies inversely with the amount of ferritin already present in the duodenal epithelial cells, which in turn reflects total body iron stores. Iron absorption by the gut also increases when there is increased normoblastic activity. (The mechanism remains unknown; the latest stuff Blood 96: 4020, 2000.) Absorption is mildly increased in hemolytic anemias or after hemorrhage. Absorption is more markedly increased in "ineffective erythropoiesis", notably in severe sideroblastic anemia and thalassemias. Huge doses of dietary iron can override the regulatory mechanism.
Iron atoms are slowly released into the plasma, where they are bound to the globulin transferrin. The amount of transferrin present is also regulated, so that more will be present when more iron is required. The iron is then carried where it is needed. (* Transferrin will only carry ferric iron, while most of the other forms of iron are ferrous.) Likewise, when a red cell (or any other cell) is destroyed, its iron is carried away by transferrin. Extra iron is stored, mostly in the liver and bone marrow. In health, it is available for incorporation into RBC's or for transport by transferrin should a shortage develop.
Storage iron (somewhere in the range of 1 gm in most people) exists in two principal forms. (1) FERRITIN is a bit of iron at the center of a protein micelle. The protein shell explains the negative Prussian blue stain. This is the short-term storage form. It is the form found, for example, in bone marrow when it is soon to be incorporated into new RBC's. (2) HEMOSIDERIN is aggregates of ferritin with much of the protein gone. This is "Prussian blue positive" iron. It is a less labile storage form that accumulates when there is excess ferritin. (* "Hemosiderin" is an archaic name chosen by von Recklinghausen. "Exogenous hemosiderin" is an iron-protein complex that forms around sites of iron injection and foreign bodies composed of iron). Hemosiderin is the yellow pigment in the halo surrounding a bruise. Tip: If an injured area is pigmented yellow three months after bruising, the patient probably has iron overload.
Humans have no special mechanism for excreting excess absorbed iron. There is ordinarily a loss of 1 mg/day or so through GI and skin cell turnover and microhemorrhages into the gut and GU tracts. A typical menstrual period results in a loss of 10-20 mg of iron. During the course of a pregnancy, the fetus absorbs 500-1000 mg of iron from the mother's bloodstream. The fetus "gets priority" and often the mother becomes iron-deficient. Donating a 500 mL unit of blood removes approximately 250 mg of iron from the body.
Iron does harm to cells by generating free radicals. Primary iron storage problems are very common, under-diagnosed, easily and inexpensively detected, potentially fatal, and very treatable. And these patients are often considered hypochondriacs for many years before the diagnosis is finally made (Ann. Int. Med. 101: 707, 1984). During the early 1990's, it was finally appreciated that around 1 man in 200 is affected (NEJM 318: 1355, 1988). HEMOSIDEROSIS ("systemic siderosis", etc.): increased total body iron (as ferritin and hemosiderin), from any cause. Most of the excess iron is in the reticuloendothelial cells. Spill-over into parenchymal cells is what can cause trouble. It is now known that 13% of people carry an autosomal gene (HFE, within the HLA complex; update Arch. Int. Med. 166: 294, 2006) for excessive iron absorption via the gut and too-easy entry of iron into the liver cells. Homozygotes have worse problems; thankfully, we have finally come to recognize this as "the most common genetic disorder in populations of European ancestry": Am. J. Med. 119: 391, 2006; the vast majority of patients of Northern European stock have a C282Y mutation, and around 0.7 of people of Northern European ancestry are homozygous for the allele (update NEJM 358: 221, 2008; of these, one man in 4, and one woman in 10 is sick from it). H63D is common worldwide. Depending on diet and iron losses, these people may or may not express their PRIMARY HEMOSIDEROSIS.
HEMOCHROMATOSIS is hemosiderosis that has damaged parenchymal cells. Total body iron stores in excess of ten gm are very dangerous. In classic cases, total body iron stores often exceed 100 gm. PRIMARY HEMOCHROMATOSIS (formerly "idiopathic", now "familial", "genetic", "hereditary", or "HLA-linked"): in which the problem is greatly increased absorption of iron from the gut. These are "primary hemosiderosis" people in which the iron overload causes illness. Perhaps 1 man in 200 will be symptomatic with this during life, and thankfully we are diagnosing it more often. Review Am. Fam. Phys. 65: 853, 2002; Lancet 370: 1855, 2007. Update on screening (from the USPSTF): Ann. Int. Med. 145: 204, 2006. So do you screen using transferrin saturation (which is awfully sensitive and not very specific), the HFE gene (which is too expensive), or a serum ferritin (which picks up the people at risk for cirrhosis, albeit late, and is also up in acute or ongoing liver abuse/disease)? The case for ferritin: Blood 111: 3373, 2008. Expect continued disagreement.
SECONDARY HEMOCHROMATOSIS most often occurs in thalassemia major and in severe sideroblastic anemias. (Before it's symptomatic, of course, it's "secondary hemosiderosiss"). In these conditions, there is greatly increased iron absorption through the gut, and the patient requires many blood transfusions with no way of disposing of the iron load. Obviously you cannot treat these patients by bleeding (why not?) Deferoxamine is life-saving in thalassemia major: NEJM 331: 567, 1994.
So, in primary hemosiderosis and hemochromatosis, the problem is that iron is absorbed too easily through the gut. A patient homozygous for primary hemochromatosis who donates blood every eight weeks will remain iron-depleted.... The tendency is encoded at HFE (Proc. Nat. Acad. Sci. 94: 2534, 1997 review, was HLA-H), discovered in 1996 in the HLA complex on chromosome 6, very closely linked to HLA-A and much like it; mouse hemochromatosis is caused by a bad beta2-microglobulin gene (Proc. Nat. Acad. Sci. 93: 1529, 1996) while the abnormal HLA-H doesn't bind properly to its microglobulin component. (* HLA-A3, B7, and B14 are often present, but the defect is linked with the chromosomes. Almost all B14 owners have the hemochromatosis gene, which is one reason (out of three) that your lecturer is confident that he carries it.) The frequency of the allele is about 14 out of every 100 chromosome 6's. Iron losses due to menstruation and pregnancies prevent expression primary hemochromatosis from ever developing in most women with the gene(s). Primary hemochromatosis is diagnosed nine times more frequently in men.
The classic hemochromatosis triad is liver trouble, diabetes mellitus, and skin discoloration. Today's list of major problems also includes cardiac rhythm disturbances, cardiac pump failure, and loss of sexuality. The liver may be enlarged on physical exam, or transaminases may be a bit high (South Med. J. 83: 1277, 1990). Around half of identified hemochromatosis patients develop overt diabetes mellitus. Since the 1990's, it has been considered good practice to screen everybody for iron overload (Gastroenterology 107: 453, 1994.) Most get the peculiar skin discoloration to some degree. The classic form of the disease is usually fully expressed around age 40-60 years. Patients, however, say their ill-health began during their 20's (Am. Fam. Physician 29: 55, March 1984).
LIVER DISEASE is the most serious problem in the majority of diagnosed hemochromatosis patients. The hepatocyte lysosomes and mitochondria are packed, and total liver iron stores are often more than one hundred times normal. There is often extensive fibrosis prior to the onset of symptoms (Arch. Int. Med. 166: 294, 2006), helping explain the classic observation that the liver enlarges even before cirrhosis develops. (The fibrosis reverses on iron removal unless cirrhosis is fully-developed.) The radiologist may notice it is unusually radio-dense. Micronodular cirrhosis (scarring that ruins the architecture of each lobule) is usually present by the time the diagnosis is made. When caused by hemochromatosis, this is called "pigment cirrhosis"). For the degree of fibrosis, the liver works surprisingly well, and clinical manifestations of cirrhosis are less severe than in alcoholic cirrhosis. However, cirrhosis is general considered irreversible and will finally kill the patient unless something else does first. (And something else usually does; only 25% of patients diagnosed to have primary hemochromatosis die of cirrhosis.) Once cirrhosis due to hemochromatosis has developed, the patient is at great risk for hepatocellular carcinoma (and to a lesser extent cholangiocarcinoma; histology Am. J. Clin. Path. 116: 738, 2001; hepatocellular carcinoma in iron overload without cirrhosis Am. J. Med. Sci. 334: 228, 2007). This is fatal and kills another 30% of patients diagnosed to have primary hemochromatosis.
Although more iron is deposited in the pancreatic acinar cells than in the ISLETS OF LANGERHANS, around 50% of patients have enough damage to their beta cells to develop symptomatic glucose intolerance.
CARDIAC INJURY is also caused by iron overload. Many hemochromatosis patients have pump failure and/or rhythm disturbances, either of which can be disabling or fatal. This is the other leading cause of death in iron-overloaded people. (How many of these deaths are assumed to be due to some other disease process? No one knows.)
ENDOCRINE INJURY is an additional problem. Patients are commonly troubled first by loss of libido, and eventually lose their secondary sex characteristics. Testicular atrophy secondary to pituitary failure with deposition in anterior lobe and Leydig cells too. It is quite reversible. Loss of testosterone in both men and women is now considered the explanation for the osteoporosis that develops in hemochromatosis patients (Ann. Int. Med. 110: 430, 1989). In addition, the adrenals, thyroid, and parathyroids are likely to be damaged, with various endocrine insufficiency syndromes. (* The role of "melanocyte stimulating hormone" in "bronze diabetes" is dubious.)
JOINT INJURY caused by iron overload (Arth. Rheum. 63: 286, 2011) is yet another major problem. Iron deposition in synovium results in synovial hyperplasia and erosion of bone and cartilage, eventually ruining the joint. In hemochromatosis, this usually affects the fingers, and is a problem for 50% of patients. In addition, the knees (and other weight-bearing joints) of hemochromatosis victims occasionally get accumulations of pyrophosphate crystals ("pseudogout", chondrocalcinosis).
SKIN PIGMENTATION in hemochromatosis is primarily due to increased melanin. (* Iron inhibits the enzyme that normally breaks down melanin.) Melanin imparts the "bronze" color; if there is enough hemosiderin in the skin, the combination looks "slate-gray".
Sepsis: For some unknown reason, Vibrio vulnificans (a raw seafood bug), Pasteurella pseudotuberculosis and Yersinia enterocolitica are much more pathogenic in the presence of iron overload. In 2011, the death of a CDC researcher from the relatively nonvirulent of plague with which he was working was attributed to his hemochromatosis (yes, they missed it even in a CDC worker -- NEJM 364: 2563, 2011). Even E. coli may get a boost in hemochromatosis (Am. J. Med. 87(3N): 40N, 1989).
You'll learn to make the diagnosis soon. In primary iron storage disease, it is important to make the diagnosis early. By the time the patient is obviously sick with hemochromatosis, he is 40 years old, has at least 20 grams of iron stored, has cirrhosis, and will probably be dead within ten years (though you can still help). If you find the tendency to accumulate iron, do this:
* "Why don't you give an iron chelator instead?" Retinal damage from deferoxamine is supposedly reversible.... Some new iron chelator drugs are available now, but I'd still stick with the no-drug approach.
Secondary hemosiderosis and hemochromatosis: Nowadays, there is a tendency to transfuse sickle cell patients heavily in the hope of preventing stroke. Patients with severe thalassemia, Blackfan-Diamond, aplastic anemia and some of the myelodysplastic syndromes also become transfsion-dependent. The abnormal anatomy is similar to primary iron storage disorders, but more of the iron is usually in the Kupffer cells, not the hepatocytes until quite late. (People who overdo oral iron and make themselves sick tend to have a lot of hemosiderin in both hepatocytes and Kupffer cells.) Hemosiderosis due to red cell transfusions ("transfusional" or "iatrogenic siderosis") is unavoidable in patients with severe anemias of decreased production unless chelators are used (and these work well: Gastroent. 141: 1202, 1211). Remember, 100 red cell transfusions delivers 25 gm of iron! This is more than enough to produce hemochromatosis. These patients are now being treated with the new iron chelator drugs (update NEJM 364: 146, 2011) which are fairly well tolerated.
* Future pathologists: If you see lots of iron in both the Kupffer cells and the macrophages of the portal area, the patient has for whatever reason been taking a great deal of oral iron and is probably otherwise healthy, though they should stop the oral iron.
Hemosiderosis due to chronic alcohol abuse results from increased absorption of iron through the gut. The mechanisms are obscure, and this fact makes it hard to distinguish pigment cirrhosis from alcoholic cirrhosis. A few old-timers blame iron-rich wine for hemosiderosis in alcoholics. (A few wines are adulterated with iron, with up to 50 mg/mL. Most have much less). See Arch. Int. Med. 112: 184, 1963. Of course, if a hemochromatosis patient drinks heavily, the liver is doomed (Gastroent. 122: 281, 2002).
Vitamin-and-mineral faddists can occasionally make themselves chronically sick by iron-overloading themselves. (This is difficult to do and probably requires the genetic predisposition. See J. Roy. Soc. Med. 77: 690, 1984.) The Bantu people ingest 100 mg or more of iron daily from beer brewed in "iron pots" (really, discarded steel oil-drums). They tend to get hemosiderosis ("Bantu siderosis"), and a few get pigment cirrhosis. (Again, genes must be important; we now know that a Bantu beer-drinker with one dose of the hemochromatosis gene will probably get hemochromatosis; NEJM 326: 95, 1992.)
You'll screen for common hemochromatosis by checking the transferrin saturation levels, and confirm by serum ferritin levels. The current cutoffs ("transferrin saturation over 45% for women or 50% for men") probably miss too many people (Am. J. Med. 120: 999, e1-7, 2007). The actual iron assays on liver biopsy tissue will probably be replaced soon by MRI (Lancet 363: 341 & 357, 2004).
Hemosiderosis due to increased or ineffective erythropoiesis ("hematopoietic siderosis", typically when there is longstanding hemolysis), mild hemosiderosis is usual because of increased absorption of iron through the gut. In the severe thalassemias and sideroblastic anemias, red cell transfusions are required, erythropoiesis is ineffective, and absorption of iron through the gut is greatly increased. The iron overload progresses to fatal hemochromatosis. These are the patients who are most often treated experimentally with iron chelator drugs.
PORPHYRIA CUTANEA TARDA is a hereditary partial defect of uroporphyrin decarboxylase that is expressed best in the presence of iron overload. You'll encounter this if you keep your eyes open! Update Blood 95: 1565, 2000.
You'll diagnose hemochromatosis by labs, and confirm with genetic testing. Liver biopsy is still handy for judging the severity (Am. J. Clin. Path. 118: 73, 2002).
Here's a summary of the genes that are understood so far (Blood 106: 3710, 2005)
TFR2: Transferrin receptor 2. Hemochromatosis type 3. The protein is expressed by hepatocytes and interacts with transferrin. It's believed to sense the body's overall transferrin saturation. Mutation causes an uncommon illness identical to common HFE hemochromatosis but on the average more severe and earlier. It's also believed that a healthy TFR2 is required in order to increase hepcidin levels when required. Gastroent. 122: 1295, 2002.
HAMP: The gene for HEPCIDIN, expressed by liver and striated muscle. Hemochromatosis type 2B. The circulating hormone hepcidin controls absorption of iron through the duodenum and its release from macrophages. Deficiency is rare but causes a severe juvenile hemochromatosis. Hepcidin: Nat. Genet. 33: 21, 2003. Hepcidin rises in iron deficiency, falls in the iron-replete in health. It's inappropriately high in anemia of chronic disease (cytokine effects on the liver) and ineffective erythropoiesis (beta-thal major), promoting iron overload; exactly how it ties into common hemochromatosis is still being worked out.
HJV / HFE2: The gene for HEMOJUVELIN. This hormone binds to the protein NEOGENIN. Hemochromatosis type 2A. This seems to be the another hormone that inhibits absorption of iron through the duodenum. When normal, it's lowered by anemia, hypoxia, and iron deficiency, permitting a higher rate of iron absorption. Nat. Genet. 36: 77, 2004; mouse J. Clin. Inv. 115: 2187, 2005; hemojuvelin senses the amount of iron in the diet overall J. Clin Inv. 115 2180, 2005.
SLC40A1 (was SLC11A3): The gene for FERROPORTIN, which is the hepcidin receptor. Hemochromatosis type 4. Another protein expressed throughout the body. It seems to permit the exit of iron from cells -- its inhibition by hepcidin from angry macrophages and IL6-stimulated liver is the basis for anemia of chronic disease. Mutions here produce a dominant hemochromatosis with iron-overloading of body macrophages but not parenchymal cells, and normal or low transferrin saturation, and usually not the classic findings of hemochromatosis unless there is another gene. They do not give up their iron on phlebotomy, but develop "anemia of chronic disease". Autosomal dominant hemochromatosis ("ferroportin disease") is in fact most often due to deficiency in ferroportin (J. Clin. Inf. 108: 512 & 619, 2001; Blood 100: 692, 2002; Blood 106: 1092, 2005' update Gastroenterology 140: 1199, 2011).
CERULOPLASMIN: Aceruloplasminemia Gut 47: 858, 2000 (hepatocytes and Kupffer cells involved uniformly, no fibrosis, neurologic disease rather than liver failure; this is NOT Wilson's).
WILSON'S DISEASE ("hepatolenticular degeneration"; Mayo Clin. Proc. 78: 1126, 2003; Gastroent. 125: 1868, 2003; Lancet 369: 379, 2007)
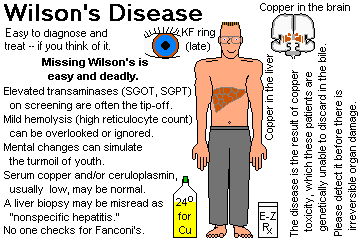
Rare but very, very important. Don't miss this diagnosis. Untreated, it is lethal. Treated, it's harmless. You'll never diagnose it unless you think of it. And it ALWAYS masquerades as something else.
Wilson's disease is an autosomal-recessive problem in which the liver is unable to dispose of excess dietary copper via the bile.
The gene was cloned in 1997, and named ATP7B; it is (no surprise) a copper-transporting ATP-ase (Am. J. Hum. Genet. 61: 317, 1997).
Serum levels of ceruloplasmin, the copper transport protein, are often (but not always) low in Wilson's. This is probably because ceruloplasmin is ordinarily sent out of the liver cells bound to copper, and if it's unbound, it is degraded too rapidly.
More helpful for your workup, serum copper, not properly carried, spills copiously into the urine in Wilson's disease. This represents the only route of copper excretion, and it is inadequate.
Eventually, the copper accumulates, damaging liver, joints, brain (especially basal ganglia), proximal renal tubule (causing wasting of solutes) and red cells (mild ongoing hemolysis is the rule), and making the distinctive Keyser-Fleischer corneal ring (you probably won't see them without a slit lamp).
The copper itself probably damages the cells in which it accumulates, maybe by free radicals or inhibiting enzymes. * Future pathologists: Stain for copper using rhodaNine or rubeanic acid!
The histology in the liver passes through fatty change to a histopathology basically identical to alcoholic hepatitis to chronic hepatitis to micronodular cirrhosis to post-necrotic cirrhosis. At autopsy, you see an unforgettable bluish-coppery colored liver, and the same discoloration in the basal ganglia.
The treatment, of course, is to remove the copper with a metal chelator. Penicillamine works wonders, but it won't cure cirrhosis or restore dead neurons.
Lab diagnosis is treacherous. Serum ceruloplasmin is low more often than not, but this is a notoriously poor screening test. Staining the liver itself for copper is treacherous. Even assaying liver tissue for copper ("normal is less than 55 micrograms/gm of liver; Wilson's has more than 250") is unreliable because you may have sampled a regenerative nodule that's copper-poor, and copper is deposited near the portal areas whenever there is chronic cholestasis. I recommend a urinary copper assay.
Missing the diagnosis of Wilson's disease clinically is still a common blunder, especially when it presents as "mental illness" (update Gut 56: 115, 2007). The physician's mistake WILL cost the patient his/her long-term health or even life.
{00018} Wilson's
![]() Wilson's disease
Wilson's disease
Joel K. Greenson MD
U. of Michigan
HEPATIC AMYLOIDOSIS: We cover this in "immuno". Amyloid accumulates in the vessels and space of Disse, and can eventually give a large, very firm liver that usually still functions acceptably. Past concerns about biopsying the liver in suspected amyloidosis seem to be groundless (Medicine 82: 291, 2003).
{53548} amyloid in the liver
![]() Amyloidosis of the liver
Amyloidosis of the liver
Joel K. Greenson MD
U. of Michigan
LIVER POISONS
A host of drugs-poisons produce typical reactions in the liver. These range from predictable ("Take enough 'Tylenol' at once and your liver cells all die") to highly idiosyncratic ("Take 'Halothane' anesthesia a second time and there's a tiny chance that sensitivity will kill you.") "Inflammation" may mimic acute or chronic hepatitis.
Acetaminophen ("Tylenol")... Massive hepatic necrosis
Allopurinol... Granulomas (* infamous for looking like the ring granulomas of Q-fever)
Alpha-methyldopa... Inflammation, granulomas, massive necrosis (idiosyncratic)
Amiodarone... Inflammation, "alcoholic hepatitis" mimic, cirrhosis (idiosyncratic); kupffer cells loaded with phospholipid ("phospholipidosis")
Anabolic steroids... Cholestasis (at least)
Azathioprine... Fatty change, necrosis and regeneration in the central zones (Arch. Path. Lab. Med. 136: 618, 2012)
Carbamazepine... Granulomas
Chlorpromazine... Cholestasis (idiosyncratic)
Diltiazem... Granulomas
Estrogens... Cholestasis (idiosyncratic), thrombosis (idiosyncratic)
Ethanol... Fatty change, alcoholic hepatitis, cirrhosis
Fenfluramine... Massive necrosis (idiosyncratic; a dozen Japanese find out what was really in their Red Chinese holistic-wholesome weight-loss pills beside lotus leaves and chrysanthemum petals: Ann. Int. Med. 139: 488, 2003)
Halothane... Massive hepatic necrosis (idiosyncratic)
{24392} "Halothane hepatitis" case; simply massive necrosis
* Hydralazine... Granulomas
* Hydrazine (a quack cancer remedy)... liver necrosis (Ann. Int. Med. 133: 877, 2000).
Isoniazid... Looks like viral hepatitis (idiosyncratic) (especially >age 35)
*7nbsp;Lapitinib... for HER+ breast cancer; people with a certain HLA get liver toxicity.
* Lithium ... Granulomas
Methotrexate... Necro-inflammation, cirrhosis (sometimes even at "safe" doses)
Oxyphenisatin (laxative)... Inflammation ("lupoid hepatitis") (who'd abuse THAT?)
* Paraquat: Hepatocellular injury that resolves (early); necrosis of the bile duct cells (late)
Pennyroyal ("holistic herbal tonic" / amateur abortifacient) ... simulates viral hepatitis (update on poisoning from Mayo's: Acad. Emerg. Med. 10: 1024, 2003)
* Phenylbutazone... Granulomas (who still uses THAT?)
* Quinidine... Granulomas
* Rituximab... Cholestatic hepatitis
* Sulfa drugs... Granulomas
Tetracycline (outdated)... Microvesicular fatty change (get rid of yours!)
* Total parenteral nutrition... Fatty change (nobody knows why; this and malnutrition are the major illnesses in which fatty change is likely to be periportal rather than central; Gastroent. 104: 286, 1993)
Valproic acid: Mixed hepatitis-like and cholestatic, or just fatty change
* Venlafaxine (antidepressant): Cholestatic hepatitis (Am. J. Surg. Path. 36: 1724, 2012).
* Future physicians: Whether or not a drug is dangerous to the liver is decided nowaday's by HY'S LAW. Hy Zimmerman's criteria: (1) AST / ALT go up to at least 3x the upper limit of the reference range; (2) and when the AST / ALT go up, at least some folks also have bilirubin go up to 2x the upper limit of the reference range without also raising alk phos (which would indicate it is only cholestasis; (3) and there's no other reason (viral hepatitis, medication). If Gilbert's is present, it can make it harder to interpret Hy's Law (Clin. Pharm. 91: 647, 2012).
REYE'S SYNDROME (Am. Fam. Phys. 50(7): 1491, 1994; NEJM 340: 1423, 1999; NEJM 340: 1377, 1999)
This poorly-understood, dread entity is an acute illness that follows another viral illness, usually 'flu or chicken pox, usually in a child or young teen.
The biphasic clinical story is typical. The pathology is, too. You will see:
The increased intracranial pressure produces vomiting. Later, in severe cases, liver enzymes rise in the serum, and then the liver itself fails. The patient may die, or be left mildly or moderately brain-damaged.
The pathogenesis of classic Reye's was never really worked out.
Several inborn errors of metabolism (notably in urea-genesis, ketogenesis, branched amino-acid metabolism, and uncoupling of oxidative phosphorylation) produce crises similar to Reye's. Studies of patients with Reye's have showed a plethora of abnormal compounds around their bodies. See JAMA 260: 3167, 1988; also Enzymes 45: 209, 1991.
Reye's, and only true Reye's, gives spectacular edema of the mitochondria, visible by electron microscopy.
In 1986, the public was strongly warned not to give aspirin to folks under age 18 except when it's specifically helpful (Kawasaki's, juvenile rheumatoid arthritis). As a result, true Reye's has nearly vanished.
"Reye's" cases nowadays are most likely to be children with decompensated metabolic defects.
Skeptics or no, "the animal model" for Reye's is a rat, which must be given both aspirin and several low doses of endotoxin (Metabolism 38: 73, 1989).
![]() Reye's Liver
Reye's Liver
CDC
Wikimedia Commons
PEDIATRIC LIVER DISEASE
BILIARY ATRESIA ("EHBA" means "extrahepatic biliary atreisa"; usually there's destruction of bile ducts both outside and inside the liver; update Lancet 374: 1704, 2009)
A catastrophic birth defect in which the common bile duct and/or hepatic ducts ("extrahepatic") and/or many larger intra-hepatic ducts are without lumens along some or all of their length. In around 90%, the entire external biliary tree is obstructed. The ducts appear to become inflamed and are destroyed before birth.
Sometimes this occurs with syndromes. More often, there's only mild inflammation and replacement of the involved areas with granulation tissue, perhaps from an intrauterine viral infection. Over time, the lesion progresses, and eventually the large bile ducts (extrahepatic, then intrahepatic) become replaced by granulation tissue (Mayo Clin. Proc. 73: 90, 1998. (* Sounds like a slow virus?)
These children (most often girls) suffer severe neonatal hyperbilirubinemia. Unless surgery can correct the problem early, the disease will progress to biliary cirrhosis, liver failure, and the need for a transplant.
If you perform liver biopsy on the infant, all you'll probably see is the ductular reaction. The bile ducts in the portal areas are generally normal especially at birth.
* A rare illness in which the intrahepatic ducts disappear in childhood is caused by defective villin, a cytoskeletal protein that maintains the microvilli in the canaliculi (Lancet 362: 1112, 2003).
NEONATAL HEPATITIS ("giant cell hepatitis")
Ongoing liver cell destruction in a baby. As in adult hepatitis, the normal liver cords are hard to see.
This has a very long differential diagnosis. It includes:
![]() Herpes hepatitis
Herpes hepatitis
Joel K. Greenson MD
U. of Michigan
Regardless of cause, the histology tends to be similar in babies.
Look for the lobular disarray, focal cell necrosis, cholestasis, and Kupffer cell hyperplasia you'd see in any kind of hepatitis.
In addition, the infant's liver regenerates well, producing prominent GIANT HEPATOCYTES with many nuclei.
Don't expect much inflammation. If you do see inflammatory cells, think of infection; however, even with infection, their absence means nothing.
{38758} neonatal hepatitis, gross
{38761} neonatal hepatitis, microscopic; with giant cells and bile stasis
Liver biopsy may show distinctive features (i.e., inclusions![]() in herpes and CMV,
basophilic smudge nuclei in
adenovirus
in herpes and CMV,
basophilic smudge nuclei in
adenovirus![]() infection, organisms
in toxoplasmosis
infection, organisms
in toxoplasmosis![]() ,
hyaline globs in alpha-1 antitrypsin deficiency.) Don't expect these always to be present.
,
hyaline globs in alpha-1 antitrypsin deficiency.) Don't expect these always to be present.
{46222} herpes simplex hepatitis, gross; trust me; all that
you can appreciate is severe injury
{46223} herpes simples hepatitis, microscopic (note the cell with two nuclei and herpes intranuclear
inclusions)
Worth mentioning again here: cirrhosis can result from galactosemia (kids) or alpha-1 antitrypsin (kids or adults).
{20137} cirrhosis, galactosemia (* future pathologists: Note the "flowers" formed by hepatocytes in this
condition)
{13301} cirrhosis, alpha-1 antitrypsin deficiency (trust me)
{13302} cirrhosis, alpha-1 antitrypsin deficiency (trust me)
{13304} cirrhosis, alpha-1 antitrypsin deficiency; PAS stain shows alpha-1 antitrypsin
granules well
As a rule, the prognosis is much more favorable than in biliary atresia. Nevertheless, a few of these children do die.
LIVER TRANSPLANT PATHOLOGY (update J. Clin. Path. 63: 47, 2010)
* HLA-matching of donor and recipient is of relatively little importance in liver transplantation, and some centers don't even bother.
You remember that HYPERACUTE REJECTION results from the presence of pre-existing antibodies (for livers, almost always ABO blood group). It's obvious at once, and the transplant must be removed. Thankfully, this almost never happens.
PRESERVATION AND REPERFUSION INJURY is especially common if the liver has been on ice for more than 24 hours. There will be extensive necrosis and neutrophilic inflammation.
* LIPOPELIOSIS results when the acquired liver was fatty (perhaps the donor was shot in a bar-fight) -- around 10% of donor livers are. Fat from the donor liver bursts out of the cells and fills the sinusoids. This self-heals nicely.
HEPATIC ARTERY THROMBOSIS is less common nowadays, but still happens. Since the bile ducts get their blood primarily from the hepatic arterial system, they take the most damage.
ACUTE CELLULAR REJECTION, usually seen in the first six months, has similar features as in other organs:
The Banff "rejection activity index" gives a score from 0-9, the sum of 0-3 for each of the above
BILE DUCT STENOSIS can develop late after surgery (scar contracts). The clue is likely to be some neutrophils and edema in the portal areas (why?)
ACUTE HUMORAL REJECTION is uncommon in the liver, supposedly because the Kupffer cells eat antigen-antibody complexes. Staining for C4d is helpful here as everyplace else.
RECURRENCE OF THE ORIGINAL DISEASE: Be it hepatitis B, hepatitis C, autoimmune hepatitis (Dig. Dis. Sci. 57: 2248, 2012), primary biliary cirrhosis, or sclerosing cholangitis -- at least you know you've bought the patient many good years.
* EPSTEIN-BARR VIRUS HEPATITIS and CMV HEPATITIS happen in transplants because of immunosuppression.
CHRONIC REJECTION usually includes the CHRONIC ALLOGRAFT VASCULOPATHY, as in other organs. There's a concentric fibrosis of the vascular intima, and some fibrosis in the parenchyma. This seldom causes the transplants to fail.
BENIGN LIVER MASSES
|
|
|
HAMARTOMAS
CAVERNOUS HEMANGIOMAS are little birthmarks, seen in maybe 1% of livers. If you see them at surgery, don't biopsy them. They used to give high-resolution scanners fits: Am. J. Roent. 162: 1113, 1994.
* Occasionally a giant hemangioma may need to be resected.
* There are several pediatric tumors of vascular origin that are difficult to prognosticate. Leave these to the pathologists.
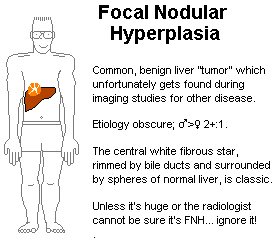
FOCAL NODULAR HYPERPLASIA (World J. Surg. 19: 13, 1995) is a curious lesion that looks like a chunk of a cirrhotic liver grafted onto a healthy one. There is usually a central star-shaped large scar.
* Future pathologists: The call is tricky, especially if you didn't get a good history. In FNH (unlike cirrhosis), look for some bile ductular change next to the fibrous bands.
* NODULAR REGENERATIVE HYPERPLASIA is a picturesque lesion in which the entire liver becomes nodular, but without fibrosis. The cause is damage to the small portal veins, as from bone marrow or kidney transplantation (nobody really knows why), thioguanine (famous; Arch. Path. Lab. Med. 128: 2004), or some other cause of portal hypertension without cirrhosis. "Some cases are idiopathic."
* MESENCHYMAL HAMARTOMA is a mix of myxoid mesenchyme and bile ducts. It is a benign-but-potentially-troublesome pediatric lesion with a trademark chromosomal marker (Cancer 74: 1237, 1994; update Arch. Path. Lab. Med. 130: 1216 & 1567, 2006).
BILE DUCT HAMARTOMAS are polka-dot malformations, mere autopsy curiosities ("Meyenburg complexes"). I'm asking you to learn them because they're quite common. There's no central necrosis and because of this, surgeons seldom mistake them for cancer at surgery.
* BILE DUCT ADENOMAS amd BILIARY CYSTADENOMAS (which can give rise to biliary cystadenocarcinomas) are uncommon. Leave the diagnosis to us.
CYSTS are bounded by biliary epithelium. In CAROLI'S DISEASE, some of the bile ducts are dilated (and thus, more prone to stasis and infection, and for some reason cholangiocarcinoma).
POLYCYSTIC LIVER may occur with either mendelian polycystic kidney disease, or as a distinct autosomal dominant or sporadic disease (Hepatol. 23: 249, 1996; Kid. Int. 76: 803, 2009).
* There is also an autosomal dominant form (gene PRKCSH Nat. Genet. 33: 345, 2003; Am. J. Hum. Genet. 72: 691, 2003; Another gene SEC63: Nat. Genet. 36: 575, 2004). Yet another is a variant of the autosomal recessive polycystic kidney disease gene: Medicine 85: 1, 2006.
If patients are uncomfortable, the cysts can be aspirated and a sclerosing agent injected. One happy patient was relieved of an extra 4200 mL of fluid (Dig. Dis. Sci. 53: 2251, 2008).
|
|
LIVER CELL ADENOMAS may occur sporadically, but are most common in women on birth control pills and men on anabolic steroids, and these are considered somehow etiologic.
Grossly, adenomas present as soft masses. The histology resembles normal liver, minus bile ducts and good lobular architecture. If you see lone arteries, it's probably an adenoma (why?) There may be cholestasis. Around a quarter are multiple (syndrome: Mayo Clin. Proc. 71: 478, 1996).
Usually liver cell adenomas cause no problems. The lesions are likely to regress when the exogenous hormones are removed. They seldom turn malignant, though occasionally this happens (Arch. Surg. 129: 712, 1994).
* Not surprisingly, patients with mutated hepatocyte nuclear factor 1-alpha, the gene for maturity onset diabetes of the young type 3, get a lot of hepatic adenomas (Gastroenterology 125: 1470, 2003). This is the only familial syndrome I've heard of.
* A paraneoplastic syndrome is pseudo-iron deficiency anemia from hepcidin production by hepatic adenomas (Blood 100: 3776, 2002). Hepcidin prevents iron from leaving macrophages and entering young erythrocytes.
* Future pathologists: Telling hepatocellular adenoma from hepatocellular carcinoma on biopsy can be tricky. One group recommends glypican-3 stain (adenomas do not stain; hepatocellular carcinomas and some nasty cirrhotic nodules usually stain: Arch. Path. Lab. Med. 132: 1723, 2008).
PRIMARY CANCERS OF THE LIVER (i.e., the hepatocytes, the intrahepatic bile ducts, and the sinusoidal lining cells)
|
 Mickey Mantle |
In the poor nations where HBV is endemic and typically transmitted mother-to-child, hepatocellular carcinoma is easily the most common cancer in men (at extra risk both because of higher HBV carriage and more body iron), and second only to cervical cancer in women. (Watch lung cancer rise to first for both sexes in the next decades.)
The level of virus in the blood correlates very strongly with cancer risk: JAMA 295: 65, 2006.
We now know that hepatitis C virus is also a very important cause, though again we don't know how (Cancer 73: 2253, 1994, still valid; update on hepatitis C-related hepatocellular carcinomas Dig. Dis. Sci. 59: 192, 2014). One thing we do know is that hepatitis B and C viruses are much more likely to produce hepatocellular carcinoma when they cause rapid, prolonged cell turnover, i.e., making Nowell's law operate more readily (Cancer 73: 1149, 1994).
Other factors enter into the picture, as well. Iron overload strongly increases risk --
long thought to be due to "free radical generation", the effect of iron on specific
molecular pathways may also be a factor (Am. J. Path. 176: 1006, 2010). Aflatoxin (from
aspergillus![]() in moldy food) is probably responsible for the trademark p53 mutation required for epidemic African
hepatocellular carcinomas (update Gastroent. 117: 154, 1999); sporadic cancers elsewhere show different mutations in p53 (Cancer 74: 30,
1994).
in moldy food) is probably responsible for the trademark p53 mutation required for epidemic African
hepatocellular carcinomas (update Gastroent. 117: 154, 1999); sporadic cancers elsewhere show different mutations in p53 (Cancer 74: 30,
1994).
"Thorotrast" exposure places a person at risk for hepatocellular carcinoma, and anabolic steroids (from the gym) supposedly do, too.
There are several more dubious risk factors cited.
* You'll find the claim that "alcohol is a major risk factor for hepatocellular carcinoma." It's been known for years that without coexisting viral infection, the danger is minimal unless one already has alcoholic cirrhosis (Eur. J. Cancer. Pev. 10: 107, 2001). The studies that "show" alcohol to be a risk factor are retrospective, and there's a likely recall bias -- "everybody knows" that alcohol is bad for your liver. And people with hepatitis B and C are more likely to be poly-substance abusers. A German group calls the claim that alcohol alone is a major risk factor a myth: Liver 20: 312, 2000. If alcohol is a real risk factor, it is minor and abuse must be heavy, and it's common for patients to have hepatitis B and/or C and/or iron overload as well (Gastroent. 127(5 S 1): S-2 and S-87, 2004 -- also makes a link to NASH; JNCI 96: 1851, 2004; Cancer 101: 1009, 2004; JAMA 313: 2263, 2015). It is most reasonable to think that alcoholic liver damage, with ongoing cell necrosis and regeneration, is acting as a promoter.
* The same claim is of course made for smoking on the basis of retrospective studies, where the link is even weaker. Again, people with hepatitis B / C are likely to be poly-substance abusers and perhaps hence smokers; recall bias is probably also a confounder ("Everybody knows smoking causes cancer.")
* Strangely, the retrospective studies I've seen that have focused on alcohol and/or smoking do not take iron status into account. This would be easy to determine by drawing blood levels.
* Antitrypsin deficiency is another dubious risk factor ("No!": Cancer 49: 2537, 2006).
* In a child -- think hereditary tyrosinemia.
Is NASH without cirrhosis a risk factor? We don't know, but data is suggestive (Arch. Path. Lab. Med. 132: 1761, 2008).
In the U.S., many (but not all) patients have cirrhosis (from their hepatitis virus and/or iron overload), but this is less common where most victims are "healthy carriers" of hepatitis B.
Wilson's cirrhosis, lupoid cirrhosis, and primary biliary cirrhosis probably do not place a person at risk, and alcoholic cirrhosis itself is at best a minor risk factor.
Grossly, the tumor may be a single mass, or arise multicentrically. The better-differentiated the tumor, the greener it tends to be (since hepatocytes want to make bile). Hepatocellular carcinomas, unlike most carcinomas, have a great propensity to invade veins, and you will probably see this grossly and microscopically.
* Worth learning sometime:
The five carcinomas that invade veins in preference to lymphatics (pretty reliable):
The two sarcomas that invade lymphatics in preference to veins (unreliable):
Histologically, the hepatocellular carcinoma may look like nearly-normal liver, or be wildly anaplastic, or something in between. Appearances vary tremendously. Pathologists look for:
The existence of "liver cell dysplasia" is now well-established (Arch. Path. Lab. Med. 135: 704, 2011). It's distinguished from hepatocellular carcinoma primarily because there is anaplasia but not (yet) any invasion of the stroma (today's pathologists use DR/CK7 keratin immunostaining to spot invasion: Cancer 109: 915, 2007) and it has ordinary hepatic sinusoids with reasonably good reticulin. It precedes hepatocellular carcinoma and is likely to turn into it. Today's imaging techniques are supposedly able to distinguish dysplastic nodules from simple regenerative nodules in cirrhosis (Br. J. Rad. 77: 911, 2004). The pathologist may do best to say, "Cannot rule out hepatocellular carcinoma."
* By contrast, "adenomatous hyperplasia" of the liver is healthy-looking cells arranged abnormally, especially thickened trabeculae. Less worrisome.
* To tell not-yet-malignant from malignant, proliferating-cell nuclear antigen, a marker for a cell thinking about dividing, is an excellent immunostain: Cancer 73: 2259, 1994. Pathologists are also counting nucleolar organizer regions (Cancer 73: 289, 1994; neat pictures of Nowell's law in action.) If hepatitis C is the underlying disease, GPC3 immunostain positivity supposedly marks the transition to true hepatocellular carcinoma (Gastroenterology 131: 1758, 2006). Today, some people are using the combination of GPC3 (glypican 3), EZH2 (enhancer of zeste homologue), and HSP70 (heat shock protein 70) to spot transformation to malignancy (Gut 60: 967, 2011). Of course, dysplastic nodules should still have reticulin.
* Dysplastic nodules do not yet show stromal invasion, but this is often hard to determine on H&E (Am. J. Clin. Path. 137: 937, 2012). Hepatocellular carcinomas tend to stain positive for the markers "Hep Par-1" (carbamoyl phosphate), glypican-3, and MOC-31, unlike dysplasia (review Arch. Path. Lab. Med. 131: 1648, 2007). Immunostains can make the diagnosis but are not useful prognostically: J. Clin. Path. 66: 687, 2013.
Not surprisingly for a tumor that arises in this way, hepatocellular carcinomas often appear to arise multifocally (but not entirely independently; see Hum. Path. 19: 403, 1988).
* The presence of a "capsule" around a hepatocellular carcinoma means exactly nothing (Cancer 70: 45, 1992).
By contrast, following resection ("hepatectomy", a confusing term -- still the best treatment, rather than transplantation Ann. Surg. 254: 527, 2011), a key prognostic indicator is the extent of vascular invasion. Involvement of a vessel 1 cm or more from the main tumor, or invasion of a vessel with muscle in its wall, are bad prognostic indicators (Gastroent. 137: 850, 2009). Of course, the bigger the tumor and the higher the grade are also independent prognostic indicators (Ann. Surg. 249: 799, 2009). Many people are alive thanks to repeated resections (three or more are common): Surgery 154: 1038, 2013.
For the little tumors, radiofrequency ablation can provide good control (Radiology 268: 589, 2013.
* The Edmondson-Steiner grading system (1954) is of little value in predicting the behavior of a particular tumor. Further, it is famously non-reproducible from pathologist to pathologist (Arch. Path. Lab. Med. 134: 1818, 2010).
* Future pathologists:
You can use aspiration cytology to tell whether a liver nodule is cancer (Gut 53: 1356, 2004). You can't use cytology to grade: Cancer 102: 247, 2004.
Hepatitis B-belt (much of Africa and East Asia) hepatocellular carcinoma is usually thoroughly anaplastic and very aggressive: Cancer 65: 130, 1990.
* The oncofetal protein SALL4 is a marker for an aggressive "progenitor" type that may be a protein targeted by tomorrow's magic bullet therapy (NEJM 368: 2266, 2013).
* Fibrolamellar hepatocellular carcinomas are rather tame and are a different disease from common hepatocellular carcinoma. Grossly they may look like focal nodular hyperplasia. The cells are packed with mitochondria. Their "pale bodies" contain albumin and fibrinogen. See Arch. Path. Lab. Med. 128: 222, 2004; Cancer 106: 1331, 2006.
* Spindle cell hepatocellular carcinomas exhibit mesenchymal markers and have an extremely bad prognosis (Cancer 77: 51, 1996).
{18799} hepatocellular carcinoma in macronodular cirrhosis;
hepatocellular carcinomas are white
{24571} hepatocellular carcinoma in cirrhosis
{39624} hepatocellular carcinoma in cirrhosis
{39708} hepatocellular carcinoma in cirrhosis
{40330} hepatocellular carcinoma in cirrhosis
{40331} hepatocellular carcinoma histology (one of many variants)
|
|
|
|
|
|
|
|
|
|
|
You can deduce the symptoms from the behavior of the tumor, but by the time the patient knows he or she is sick, it's often too late. When a cirrhotic deteriorates rapidly, it's common to find a hepatocellular carcinoma at autopsy.
Paraneoplastic syndromes include polycythemia (erythropoietin production) and hypoglycemia (insulin-related growth factor production).
Death occurs through rupture, venous obstruction, liver failure, or (less often) lung metastases.
Without treatment, the disease is of course lethal. Nowadays, many hepatocellular carcinomas are discovered incidentally, and especially if there is no cirrhosis, the disease is often curable (Gastroenterology 122: 1609, 2002).
Surgery is often successful since since the tumor tends to metastasize (to the lungs) only late. Nowadays, even "huge" single-nodule tumors 10 cm or larger, when resected, yield a 40% cure rate (Am. J. Surg. 198: 693, 2009).
More recently, resection of the whole liver with liver transplant is becoming a mainstay of therapy. Nowadays, US surgeons resect what they can and transplant when a liver becomes available. This leaves around 3/4 of the patients alive at 5 years, and 1/2 cancer-free (Ann. Surg. 250: 738, 2009).
Since the early 1990's, good palliation has often achieved simply by embolizing the hepatic artery, since the cancer's blood supply is mostly arterial (Cancer 73: 2259, 1994). Chemotherapeutic agents on particles can be delivered in this way ("chemoembolization"). And nowadays, radiofrequency ablation or even injecting absolute alcohol under CT guidance (which gets the patient drunk) can supposedly produce cures.
* Oncologists have a variety of interesting approaches. Small tumors have been treated with microwaves (Cancer 74: 817, 1994; "residual tumor" may be non-active -- future pathologists will enjoy reading about the picturesque after-effects in Am. J. Gasto. 98: 2052, 2003) or ethanol injection (Radiology 192: 407, 1994). Both are tributes to our ability to detect tiny tumors by serum screening and scanning.
Today's standard will probably soon be a combination of chemoembolization (embolization of the hepatic arteries with particles bearing antineoplastic drugs) plus radiofrequency ablation (JAMA 299: 1669, 2008; from the new China; also Ann. Surg. 248: 617, 2008).
Still, the long-term outlook is grim without major surgery plus a lot of good luck. As with lung cancer, after non-surgical "cure" of one hepatocellular carcinoma, it is not unusual for a new clone to arise from the same premalignant cellular milieu (Nowell's law triumphant; Cancer 63: 2207, 1989).
* Sorafenib, a pricey inhibitor of Raf / VEGF / PEGF kinases, is the first molecular-target oral therapy that does any good for the incurable hepatocellular carcinoma patient (Med. Clin. N.A. 93: 885, 2009; Cancer 115: 428, 2009; AJR 194: 5-14, 2010).
My favorite pathology article from 2008 was a study that showed really good prognostication from gene expression studies on formalin-fixed tissues (NEJM 359: 1995, 2008). This is the future of surgical pathology -- enjoy it.
CHOLANGIOCARCINOMA-type cancers (see below) can occur within the liver, or as a mix with hepatocellular carcinoma.
They are desmoplastic adenocarcinomas, often mucin-positive. Of course, the cells will not secrete bile.
The best-known risk factors for this lesion are (1) "Thorotrast" exposure; (2) liver fluke infestation; (3) idiopathic sclerosing cholangitis (THE risk factor in the patients you'll see, multiplyng the base risk by maybe 1500 x); * (4) Caroli's.
* Perhaps real risks: hepatitis C infection and alcohol abuse (Cancer 115: 4564, 2009). This is supported by looking closely at the bile ducts for premalignancy.
Surgeons see Ann. Surg. 245: 755, 2007. Only the stage seems to matter for prognosis.
|
|
HEPATOBLASTOMA is a pediatric solid cancer most common under age 3. Surgical resection (followed by liver transplant, of course) is often curative.
Ask a pediatric pathologist to show you a sample. As in Wilms' tumor, there's often a variety of different kinds of epithelial and/or mesenchymal-derived structures. Alpha-fetoprotein is high, and FSH/LH often high too. (What would the latter do?)
The important prognostic factor is stage; histologic type is less important (Ped. Path. 12: 167, 1992).
Histopathologists are now talking about the "cell of origin" (obviously a stem-cell) even predating the differentiation into endoderm and mesoderm (Am. J. Path. 148: 321, 1996). Update Cancer 98: 668, 2003.
* Along with rhabdomyosarcoma, hepatoblastoma is more common in children conceived with assisted conception -- most other pediatric cancers aren't (NEJM 369: 1819, 2013).
* "Undifferentiated embryonal sarcoma", once considered a hepatoblastoma variant, is probably a different entity, though it can light up with alpha-fetoprotein, show anaplastic-looking bile ducts, and so forth.
HEPATIC (HEM)ANGIOSARCOMA, a rapidly lethal cancer, is infamous for having been caused by exposure to industrial poisons (vinyl chloride, arsenic) and the radioactive contrast medium "Thorotrast".
METASTASES TO THE LIVER need to be mentioned, too.
Liver and lung are the two organs most often involved in fatal cancer. Around 35% of patients dying of cancer have "liver mets", and about the same percent have "lung mets". The biggest livers are those bearing metastatic carcinoma.
Primary cancers of the liver (hepatocellular carcinoma, angiosarcoma, cholangiocarcinoma) often appear multiple. But you can distinguish metastatic cancer from primary liver cancer fairly easily:
Metastatic tumor in the liver tends to umbilicate (i.e., undergo central necrosis because of the watershed effect). Hepatocellular carcinomas (which are very vascular) and cholangiocarcinomas seldom do this.
If it's a cirrhotic liver, chances are it's hepatocellular carcinoma.
Here's a rule that usually works: Untreatable liver metastases portend death within twelve months.
* Some folks are living longer than the usual 12 months now thanks to cryoablation: Am. J. Surg. 171: 27, 1996.
Remember, families of patients dying with liver metastases often believe the patient died of "liver cancer", unrelated to any known primary.
Lymphomas and leukemias in the liver cluster on the portal areas. Erythroid cells in the liver cluster in the sinusoids.
|
|
|
|
|
![]() Liver metastases
Liver metastases
Primary was pancreas
Wikimedia Commons
FOR FUTURE LIVER PATHOLOGISTS AND EXAM-TAKERS:
Necrosis in zones of the liver:
ZONE 1 (periportal) is hit hardest in phosphorus poisoning, cocaine abuse and eclampsia
ZONE 2 (midzonal) is hit hardest in yellow fever
ZONE 3 (centrilobular) is hit hardest in hypoxia and shock (i.e., it gets the least oxygen), in acetaminophen toxicity, and in CCl4 toxicity. NOTE: This is the best place to look for bile plugs. Why?
Other keys to the diagnosis:
LOTS OF HEMOSIDERIN Is it located predominantly...
PRIMARY HEMOCHROMATOSIS (zone 1 is most heavily involved, then zone 2, then zone 3). If there's cirrhosis, you'll see iron in the fibrous tissue and biliary epithelium.
HEMOLYSIS (and some cases of transfusional hemochromatosis)
LONGSTANDING CONGESTION / REPETITIVE ISCHEMIA (why?)
Hint: If there is cirrhosis from most any cause, secondary iron overload is common. If the bile ducts and fibrous stroma of a cirrhotic are iron-poor, the problem is NOT primary hemochromatosis.
Chronic hepatitis with a large predominance of plasma cells: AUTOIMMUNE (LUPOID) HEPATITIS (chronic active hepatitis from any cause can look the same)
Chronic hepatitis with lots of fatty change in a non-drinker (and nothing that says NASH): HEPATITIS C
Mallory's hyaline in zone 3, plus fat and polys: ALCOHOLIC HEPATITIS / NASH (also consider ileal bypass or amiodarone toxicity)
Random confluent-lytic necrosis: UNUSUAL VIRAL INFECTIONS, IN A BABY OR IMMUNOSUPPRESSED PERSON (* CMV![]() ,
herpes
,
herpes![]() simplex
or zoster
simplex
or zoster![]() ,
adenovirus
,
adenovirus![]() , echovirus; also consider whether the biopsy was obtained during
surgery)
, echovirus; also consider whether the biopsy was obtained during
surgery)
Polys in the lumens of bile ducts: ASCENDING CHOLANGITIS
* Mallory's hyaline in zone 1: Wilson's disease, primary biliary cirrhosis, amiodarone
* Lipofuscin in Kupffer cells: A sign of recent (last few months) hepatocellular necrosis
* d-PAS positive stuff in Kupffer cells: A marker that the patient is on hyperalimentation
Polys in the portal areas, bile duct proliferation: Chronic or intermittent bile duct obstruction
Bile lakes, bile duct proliferation, edema of the portal areas: Acute bile duct obstruction.
Onion-skinning around the interlobular bile ducts (and eventual replacement by fibrous onions): Primary sclerosing cholangitis
Lots of eosinophils in the portal areas: Drugs, parasites (remember
schistosomes![]() ),
* primary biliary
cirrhosis,
* primary sclerosing cholangitis
),
* primary biliary
cirrhosis,
* primary sclerosing cholangitis
Granulomas in the liver: TB![]() , histoplasmosis (Am. J. Clin. Path. 113:
64, 2000 -- huge array of different lesions, with good granulomas being
the exception), primary biliary cirrhosis, sarcoid, drugs as above, Crohn's
disease, Hodgkin's, remote effect of Hodgkin's,
Q-fever
, histoplasmosis (Am. J. Clin. Path. 113:
64, 2000 -- huge array of different lesions, with good granulomas being
the exception), primary biliary cirrhosis, sarcoid, drugs as above, Crohn's
disease, Hodgkin's, remote effect of Hodgkin's,
Q-fever![]() (distinctive
"target" or "fibrin ring" grnaulomas),
listeria, brucella.
(distinctive
"target" or "fibrin ring" grnaulomas),
listeria, brucella.
![]() TB of the liver
TB of the liver
Great granulomas
Pittsburgh Pathology Cases
* Fibrosis around individual cells: Congenital syphilis![]()
Star-shaped portal-tract scar containing proliferating bile ducts filled with eosinophilic globs: Cystic fibrosis.
Pericellular chicken-wire scar around central vein: Central hyaline sclerosis (EtOHism / NASH)
* Fibrosis with bridging and regeneration, but no nodules: Hepatoportal sclerosis
* Fibrosis with bridging, but no nodules or regeneration: Non-cirrhotic bridging fibrosis
Cholestasis without other abnormalities: Think of drug-effect, post-surgery, sepsis, pregnancy.
Lots of neutrophils in the sinusoids: Good marker for sepsis. They are easier to see here than in the spleen.
Ground-glass hepatocytes: Those caused by hepatitis B virus infection stain positive with * orcein, * Victoria blue, and the appropriate immunostain.
* Scarring around, or loss of, portal veins in the portal areas, but everything else is okay? Idiopathic hepatoportal sclerosis! Thankfully, very uncommon.
Update on how to percuss and feel the liver (and whether it's worthwhile; it is): South. Med. J. 87: 182, 1994.
|
|
BILE DRAINAGE SYSTEM
|
|
{15774} normal gallbladder, from the outside
{15775} normal gallbladder, opened up to show mucosa
{08820} "normal gallbladder" with Rokitansky-Aschoff sinuses;
lumen and mucosa on right
{15287} normal gallbladder
Worth remembering from previous course work:
BIRTH DEFECTS
We've already looked at the dread BILIARY ATRESIA.
PHRYGIAN CAP is an anomaly of the gallbladder in which the fundus is folded over on itself. It's a radiologist's curiosity in which gallstones are more likely to form.
| * Western civilization fans: A real "phrygian cap" is the flop-over headgear worn by the French Revolution Lady allegorical figure, and on the swordpoint on the US Army seal. Smurfs wear phrygian caps. |
 |
GALLSTONES (all about them: NEJM 328: 412, 1993; laparoscopic surgeons JAMA 269: 1018, 1993; lithotripsy Gut 35: 417, 1994)
About 1 adult in 10 has gallstones. Usually these are asymptomatic for life. However, they can cause major problems. There are 600,000 gallstone surgeries in the U.S. yearly.
CHOLSTEROL STONES may be pure or contaminated with calcium, oxalate, and/or bilirubin. They result from bile becoming super-saturated with cholesterol. They are yellow and range in size from infinitesimal (in aggregate, these are the potentially-lethal "sludge") to large barrel-shape casts of the gallbladder.
You can read for yourself about "lithogenic bile", etc., etc. Bile that is merely cholesterol-supersaturated becomes liquid crystals; excess mucin production from the gall bladder that follows causes nidus formation and growing crystals (Nat. Med. 10: 1303, 2005). Harvard team on the physics of cholesterol gallstone formation: Front. Biosci. 13: 401, 2008.
Decide for yourself whether gallstones are "a disease caused by the Western diet", or whether paleontologists are correct in exhibiting dinosaurs' gallstones, or whether there may be truth in both.
* The only two gallstone loci known are UGT1A1 (Gilbert's locus but a different allele) and ABCG8 (a cholesterol processor). See Gastroentology 139: 1942, 2010.
Everybody knows "the typical cholesterol stone patient" and the "F's": fair-skinned, fat, female, fertile, flatulent, forty year old.
{00041} faceted, mostly-cholesterol stones; despite the black
bilirubinate coating, the large size and smooth faceted surfaces say "cholesterol"
is their major component
{08425} gallstone city!
{17591} gallstone city!
|
|
BILIRUBIN STONES ("mulberry stones"; "black pigment stones") are calcium bilirubinate and bilirubin polymer. They result from bile becoming super-saturated with unconjugated bilirubin, i.e., when there is hemolysis (sickle cell, hemoglobin C, spherocytosis, autoimmune Coombs-positive anemia; also remember intramedullary hemolysis, especially in the thalassemias).
{18772} bilirubin stones
Regardless of composition, gallstones present problems when they:
{10544} common duct stone
{49247} common duct stones;
common bile duct has been opened. Liver at top
{49181} gallstone ileus
{49182} gallstone ileus
ACUTE CHOLECYSTITIS (NEJM 358: 2804, 2008)
Inflammation of the gallbladder, usually containing gallstones, usually with evidence of longstanding chronic cholecystitis, and often (but not always) with a superimposed bacterial infection.
The usual bugs are E. coli or enterococci. Less common are clostridia and salmonella (typhoid is carried in the gallbladder as a biofilm on gallstones).
No one really understands the pathogenesis of acute cholecystitis, and "Big Robbins" is probably right in saying that the bacteria are secondary invaders. It is inviting to think that a stone may plug the outlet, and the gallbladder mucosa may then become infarcted due to compression by the muscularis propria's futile efforts to push the stone out. Read about "poisoning by lysolecithin", etc., etc., on your own time; it is speculative.
In the absence of gallstones ("acalculous" cholecystitis, 10% of cases), there's generally some systemic disease, and/or perhaps edema of a Reister valve obstructs the outlet. It can follow severe shock by a few days; perhaps the cause is ischemic necrosis of the biliary epithelium. It looks histologically like acute cholecystitis.
The pathology is what you'd expect. The gallbladder is red, swollen, and tense. There may be pus on the inside (even an "empyema"), fibrin all over the outside, or even gangrene of the wall.
{49242} cholecystitis, acute-on-chronic
|
|
{08822} chronic cholecystitis, histology
|
Australian Pathology Museum High-tech gross photos
|
This just means fibrosis of the gallbladder, maybe from repeated bouts of mild or severe acute cholecystitis, maybe "just from the irritation of having gallstones". There are almost always gallstones. Look for:
* "Follicular cholecystitis" refers to lymphoid follicles in the mucosa.
The "porcelain gallbladder" has undergone dystrophic calcification.
NON-PROBLEMS
"Cholesterolosis", or "strawberry gallbladder", is a pathologist's delight seen in maybe 5% of autopsies. Little clusters of cholesterol-laden macrophages develop just under the epithelium, looking like strawberry seeds. Its significance to human health is zero.
{13362} strawberry gallbladder ("cholesterolosis"), gross
{08108} strawberry gallbladder, histology; foam
cells (phagocytic macrophages) contain cholesterol
"Inflammatory polyps" are benign nubbins on the gallbladder wall.
"Mucocele" ("hydrops") is a gallbladder filled with mucus instead of bile, because its cystic duct has become obstructed. The gallbladder won't visualize on x-ray.
{38827} hydrops ("mucocele") of the gallbladder
Benign tumors of the gallbladder are uncommon, and usually incidental findings.
CARCINOMA OF THE GALLBLADDER
Adenocarcinomas of the gallbladder are uncommon (but more common than cholangiocarcinomas of the bile ducts). They are usually preceded by (and most people think caused by) gallstones. As you would expect, women are at increased risk.
* It's been claimed that chronic carrying of typhoid or paratyphoid bacteria increases the risk by about fifty times (Lancet 343: 83, 1994; somebody please take a look at this study and tell me if it's flawed.)
Usually, these have already invaded the liver by the time they become symptomatic (i.e., have obstructed the common bile duct). If they are found while still in the gallbladder mucosa, removal is curative (Am. J. Clin. Path. 135: 637, 2011).
{26327} adenocarcinoma of the gallbladder, gross
{26330} adenocarcinoma of the gallbladder
{26333} adenocarcinoma of the gallbladder
{49249} adenocarcinoma of the gallbladder
{38821} adenocarcinoma of the gallbladder, with stones
{38824} adenocarcinoma of the gallbladder, no stones
CANCER OF THE BILE DUCTS
Adenocarcinomas arising from these structures are uncommon. Of course, they produce obstructive jaundice.
Risk factors include ulcerative colitis and infestation with liver flukes or ascaris worms. Unlike for cancer of the gallbladder, men are at greater risk.
The most common site is at the ampulla of Vater, and this is often considered separately. The "Klatskin tumor" at the junction of the hepatic and common ducts is also common, but any point on the common bile duct or a hepatic duct can be the primary.
Cancer of the ampulla tends to be mucin-producing. Cancers arising elsewhere from among the bile ducts tend to be desmoplastic.
The symptomatology is what you'd expect. Itching makes these patients particularly miserable.
Future surgeons: COURVOISIER'S LAW!
In obstructive jaundice, if the gallbladder is palpable, the patient has cancer.
Explanation:
In obstructive jaundice below the level of the cystic duct, and assuming the cystic duct is patent, the pressure in the gallbladder will be high.
If the problem is a common duct stone, the gallbladder will be scarred up due to previous episodes of acute cholecystitis, or chronic cholecystitis. Therefore, it will not expand.
If the problem is cancer of the pancreas, common duct, or ampulla, the gallbladder wall itself is normal, so it will be tremendously expanded and easily palpated.
This works pretty well, but nobody's going to base a decision on it nowadays. Can you think of EXCEPTIONS? Sure you can!
To date, there is no established role for "alternative and complementary medicines" in the treatment of any liver disease, though it's possible some of the herbal remedies might have some effect. A famous and elaborate protocol by a naturopath for treatment of chronic hepatitis C, which centers around a very special recipe for breakfast museli, led to drops in transaminase levels but he made no effort to control for the fact that he also made his patients stop drinking alcohol. This invites an obvious conclusion. Plus, the risk of hepatotoxicity from herbal concoctions is well-known. See Liver Int. 23: 213, 2003; Lancet 361: 101, 2003; Med. J. Aust. 178: 442, 2003; Clin. Liver Dis. 7: 453, 2003; Clin. Tox. 46: 819, 2008 (from Hong Kong, where the problem is horrible.) In Korea, use of "alternative medicine" seems to be the most common cause of elevated liver enzymes in breast cancer patients (J. Kor. Med. Sci. 19: 397, 2004). In Oregon, "complementary herbal remedies" are now THE cause of massive hepatic necrosis requiring transplantation, exceeding even acetaminophen overdoses and all the viruses combined. (Arch. Surg. 138: 852, 2003). Even the much-touted "milk thistle" (silibindin; review Am. Fam. Phys. 72: 1285, 2005), though non-toxic, hasn't shown any robust evidence of working (Am. J. Med. 113: 506, 2002). There is a recent positive report for hepatitis C (Gastroent. 135: 1561, 2008) and another for NASH (Dig. Dis. Sci. 2387, 2007); both are small overseas studies. The pediatric oncologists at Columbia tried it for controlling the hepatotoxicity of chemotherapy; the milk-thistle group seemed to show a barely-significant benefit by measuring liver enzymes (Cancer 116: 506, 2010) -- milk thistle supposedly competes with other toxins for uptake by hepatocytes, so perhaps this is why the benefit (Eur. J. Emerg. Med. 17: 17, 2010.) Another failure, this time for hepatitis C under treatment: JAMA 308: 274, 2012.
* SLICE OF LIFE REVIEW
{08820} gall bladder, normal
{08826} liver, normal
{11798} liver, normal
{11807} liver, normal
{14874} liver, porcine
{14875} liver human, normal
{14875} liver human, normal
{14876} liver human, normal, central vein
{14876} liver human, normal
{14877} liver (sinusoids), normal
{14877} liver (sinusoids), normal
{14878} liver (sinusoids), normal, space of Disse
{14878} liver (sinusoids), normal
{14879} liver, portal triad
{14880} liver, portal triad; a=vein, b=duct
{14883} gallbladder, normal
{14885} gallbladder (epithelium), normal
{15104} liver
{15105} liver
{15106} liver
{15107} liver, pig
{15281} liver, normal
{15281} liver, normal
{15282} liver, normal
{15282} liver, normal
{15283} liver, normal
{15283} liver, normal; a=vein, b=artery, c=duct
{15284} liver, pig
{15285} liver, pig
{15286} gall bladder, normal
{15286} gall bladder, normal
{15287} gall bladder, normal
{15287} gall bladder, normal
{15288} gall bladder, normal
{15288} gall bladder, normal
{15773} gall bladder, normal
{15774} gall bladder, normal
{15775} gall bladder, normal
{15776} liver, normal
{15778} liver, normal
{16043} liver, normal in fetus
{16074} necrosis, liver junct.; normal
{17592} gallbladder, normal
{19721} gall bladder, normal
{20888} liver, portal triad
{20889} liver, portal vein
{20890} liver, hepatic artery
{20891} liver, bile duce
{20892} liver, hepatocyte plate
{20893} liver, central vein
{20894} liver, central vein
{20895} liver, portal triad
{20896} liver, portal triad and central vein
{20897} gall bladder
{26039} liver, normal
{26042} hepatocytes, normal aspirate
{26045} hepatocytes, normal aspirate
{39498} liver, normal
{39764} liver, normal
I hate liver, liver makes me quiver,
Well, maybe you just haven't had it cooked right...
Naaah, I've had that stuff sauteed, I've had it breaded and broiled and broasted and braised,
Well, what happened?
Well, I was about 9 months..my mammy gave me a hunk of that Gerber's baby food.
At nine months, what'd you say? I said...
I hate liver, liver makes me quiver,
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about
pathology of the liver
to consult these standard textbooks.
In my notes, the most helpful current
journal references are embedded in the text.
Students using these during lecture strongly prefer this.
And because the site is constantly being updated,
numbered endnotes would be unmanageable.
What's available online, and for whom, is always changing.
Most public libraries will be happy to help you get an article
that you need. Good luck on your own searches, and again,
if there is any way in which I can help you, please contact me at
scalpel_blade@yahoo.com.
No texting or chat messages, please. Ordinary e-mails are welcome.
Health and friendship!
liver makes me curl right up and die, makes me cry...
it gives you hives, gives you scurvy, turns my stomach topsy turvy,
liver just simply ain't my bag,
makes me gag, makes me want to throw up...
Now liver is neither solid or liquid, but merely an amorphous, viscous colloid of putrid
protein...
It is located between the 7th and the 10th dorsal vertebrae,
JUST south of the diaphragm, "lounging like a worm on a pillow of fat."
Now did you ever look at the word liver?
Is it any coincidence that there are 5 letters in the word liver?
The same number of letters as in the word death? the word drugs? the word hippy?...
I've had it diced and sliced and riced and sunnyside up over easy fried, and it still comes out, LIVER....
I hate liver, liver makes me quiver, liver makes me curl right up and die, (makes me cry)...
Why, first time I had it, I didn't like it at all...
Well, I rolled it around, looked at her eyeballs, spit it out and uttered my first words...
liver makes me curl right up and die, makes me cry...
it gives you hives, gives you scurvy, turns my stomach topsy turvy,
liver just simply ain't my bag.
-- "Second City", Chicago radio group, 1970's
Kanel's Atlas of Liver Pathology
Morson and Dawson's Gastrointestinal Pathology
Robbins and Cotran Pathologic Basis of Disease
Rosai and Ackerman's Surgical Pathology
Rubin's Pathology: Clinicopathologic Foundations of Medicine
Scheuer's Liver Biopsy Interpretation
Demay's Cytopathology
Sternberg's Diagnostic Surgical Pathology


| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |


 Liver / Pancreas
Liver / Pancreas
