Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
|
Cyberfriends: The help you're looking for is probably here.
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Describe what the kidneys do in health. Describe the different parts of the nephron, what each does, and what things are likely to happen when each malfunctions.
Recognize the causes of acute renal shutdown and of irreversible renal failure. Describe the many clinical and anatomic consequences of uremia.
Recall the clinical, gross, and microscopic pictures, when applicable, for each of the following:
Horseshoe kidney
Adult polycystic kidney disease
Autosomal recessive polycystic kidney disease
Acquired dialysis cystic disease ("trans-stygian kidneys")
Medullary sponge kidney
Simple cysts
Multicystic dysplastic kidney ("cystic dysplasia")
Describe what is happening in each of the following syndromes. Tell what you might see clinically, grossly (when applicable) and microscopically (when applicable), and mention its common causes.
Nephritic syndrome
Nephrotic syndrome
Rapidly progressive glomerulonephritis
Asymptomatic hematuria of glomerular origin
Hemolytic-uremic syndrome
Acute tubular necrosis
Tubular proteinuria
Fanconi syndromes
Acute pyelonephritis
Chronic interstitial nephritis (including "chronic pyelonephritis")
Diabetes insipidus
Urate, oxalate, hypokalemic, myeloma, and radiation nephropathies
Benign "essential" high blood pressure
Malignant hypertension
Renal high blood pressure
Other secondary high blood pressure syndromes
Atheroembolization
Hydronephrosis
Nephrolithiasis
Renal cell carcinoma
Wilms tumor
Transitional cell (urothelial) carcinoma of the renal pelvis
Explain how casts form. Mention the various casts that may appear in the urine in health and disease, and what they mean.
NOTE: For changes in blood chemistry (blood urea nitrogen, creatinine, creatinine clearance, etc.), see my unit on renal function testing I also have a unit on urinalysis.
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- Kidney
Lectures follow Textbook
![]() QUIZBANK...
Metabolic #'s 82-92; Kidney (all)
QUIZBANK...
Metabolic #'s 82-92; Kidney (all)
What's the difference between beer and urine?
About twenty minutes!What's the difference between a nephrologist and a neurologist?
The 'p'!
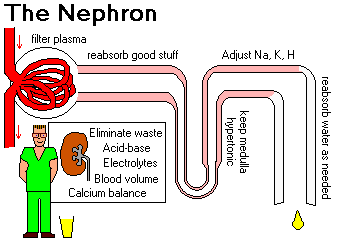
One thing that makes kidney pathology so hard is that many of the words sound alike. Here are the most troublesome words:
Collagenized glomeruli: These glomeruli have been obliterated by dense type I collagen. Most often, the collagen has been laid down concentrically on the inner surface of Bowman's capsule, as in longstanding arterial/arteriolar disease. Collagenized glomeruli are more often called hyalinized or obsolescent, despite the fact that these terms are less specific.
Diffuse: As applied to glomerular disease, all the glomeruli are involved.
Endothelialitis: Lymphocytes under the endothelium in the arterioles and venules; one hallmark of T-cell-mediated transplant rejection, but not diagnostic
Fibrosis: Dense, type I collagen deposited in the glomeruli and/or interstitium and/or vessels.
Focal: As applied to glomerular disease, some glomeruli are involved and some are not.
Global: As applied to glomerular disease, if a glomerulus is involved, all portions of it are involved.
Glomerulonephritis: As usually used, this implies that the glomeruli are sufficiently inflamed to cause at least a few of them to lose blood into the tubules.
("Glomerulonephritis" without nephritic syndrome -- i.e., "membranous glomerulonephritis", "minimal-change glomerulonephritis", etc. -- is a less-common usage. Better to call these "glomerulopathy".)
Glomerulopathy: Any primary problem with the glomeruli.
Glomerulosclerosis, diffuse: Thickening of the basement membrane as a result of diabetes mellitus.
Glomerulosclerosis, focal/segmental: A pattern of injury with foot process fusion and hyalinization of some lobules in some glomeruli. It has nothing to do with diabetes mellitus.
Glomerulosclerosis, nodular: Diabetes mellitus with Kimmelstiel-Wilson disease. Always superimposed on diffuse glomerulosclerosis.
* Hyalinosis: A distinctive, homogeneous pink blob seen in certain sick glomeruli, notably those damaged by FSGS, diabetes, or other causes of hyperfiltration.
Hyalinized glomeruli: A term that can mean collagenized or sclerotic glomeruli.
Hypernephroma: Obsolete term for renal cell carcinoma.
Nephritis: Used by itself, this means "glomerulonephritis".
Nephritis, interstitial: Inflammation of the kidney that spares the glomeruli. Includes cases formerly diagnosed as "chronic pyelonephritis". Causes U-shaped cortical scars.
Nephroblastoma: The common childhood cancer of the kidney -- Wilms tumor.
Nephrocalcinosis: Calcification of the basement membranes of the tubules in the medullae. It has nothing to do with calcium stones. A little calcification here is common, especially in older people. Extensive calcification suggests hypercalcemia and/or hyperphosphatemia ("metastatic calcification").
Nephrolithiasis: Stones (calculi) in the pelvis of a kidney
Nephropathy: Anything wrong with the kidney -- glomeruli, tubules, or vessels.
Nephrotic syndrome: The sequelae of heavy protein leakage at the glomerular capillaries.
Nephrosclerosis: Disease of the renal arteries and/or arterioles.
Nephrosclerosis, arterial: Multiple small infarcts destroying scattered groups of glomeruli. Causes V-shaped cortical scars. Usually caused by atheroembolization.
Nephrosclerosis, arteriolar: Vascular disease that destroys scattered individual nephrons. Causes pitted or sandpaper-surface kidney. "Benign nephrosclerosis". Caused by high blood pressure and/or diabetes.
Nephrosclerosis, benign: Arteriolar nephrosclerosis due to "benign essential hypertension".
Obsolescent glomeruli: Another term that can mean collagenized or sclerotic glomeruli.
Pyelonephritis: Inflammation of the interstitium of the kidney. Current usage mostly limits this to bacterial infection.
(Glomerulo)Sclerosis: As applied to kidney, this means increased basement membrane/mesangial matrix material obliterating loops of a glomerulus.
Sclerotic glomeruli: These glomeruli are fully replaced by basement membrane/mesangial matrix material, as in advanced diffuse, nodular, or focal-segmental glomerulosclerosis. They are also called hyalinized or obsolescent.
Segmental: As applied to glomerular disease, some portions of some glomeruli are involved and some other portions of the same glomeruli are spared.
Here is a list of the more important entities that are likely to be caused by a particular pattern:
 |
Subepithelial, large, irregularly-spaced ("coarse granules"; "humps")
Diffuse proliferative GN (especially post-streptococcal |
 |
Subepithelial, uniform, evenly-spaced ("fine granules evenly spaced")
Membranous glomerulopathy (any cause) Lupus, class V
|
 |
Anti-GBM diseases ("smooth linear" -- don't expect to see these on EM)
Goodpasture's, others |
|
Subendothelial (various descriptions, you will only need to recognize on EM) Membranoproliferative GN type I
Also look here for amyloid deposits.
|
 |
Intramembranous (various descriptions, depends on the disease)
Dense deposit disease (membranoproliferative GN type II)
|
 |
Mesangial ("mesangial pattern")
IgA nephropathy
|
{11850} kidney, model
{11851} kidney, model, close-up
INTRODUCTION TO KIDNEY DISEASE
To review:
In health, your body fluid tonicity is regulated by ADH and thirst. These mechanisms work extremely well.
In health, your body fluid volume is regulated primarily by atrial natriuretic peptide, which is produced when the right atrium feels that extra stretch, and which mediates a host of effects. Since body fluid tonicity is so well-regulated, extracellular body fluid volume is essentially a measure of total body sodium. This mechanism works pretty well.
In health, your total body potassium is regulated primarily by diffusion of potassium out of the near portion of the distal convoluted tubule in response to intracellular pH shifts, which in turn reflect your potassium load. This mechanism doesn't work very well. You already know that the colon helps get rid of excess potassium also. Aldosterone helps at both sites.
In health, your total body base ("the metabolic component of your pH adjustment", etc., etc.) is regulated by the carbonic anhydrase mechanism in the proximal tubule, i.e., the more CO2 on board, the more intracellular bicarbonate is produced, the more bicarbonate is resorbed, and the more protons get sent away in the urine. This mechanism works kind-of, and it takes days.
The kidney also must help regulate serum calcium, in concert with other organs (parathyroids, bone, gut).
Finally, the kidney senses anemia and produces erythropoietin as needed to encourage production of red blood cells and keep the circulating hemogobin concentration at an appropriate level.
Kidney disease is prevalent and usually serious.
In 2009 in the USA, around 100,000 people began dialysis, and 350,000 were receiving ongoing dialysis. The numbers keep increasing mostly because the population is living longer, Uncle Sam is happy to dialyze people at any age, and today's end-stage kidney has usually been taking damage for decades from hypertension, diabetes, chronic infection, and/or cystic disease. The picture is similar around the world: Lancet 365: 331, 2005.
In 2009, the US government paid $29 billion to maintain its chronic hemodialysis program. Everyone has a "right" to hemodialysis under this program, which is curious. By contrast, most other industrialized democracies ration dialysis but provide basic health care to everyone (JAMA 273: 1118 & 1123, 1996).
The typical end-stage kidney patient spends a few years on dialysis (not as bad as it once was, but still not fully healthy -- Am. J. Kid. Dis. 27: 557, 1997, annual cost to "somebody" at the time was $47,000 for peritoneal dialysis and $76,000 for hemodialysis) and perhaps gets transplanted a few times (a better way to live, and cheaper; costs are around $52,000 per transplant procedure Surg. Clin. N.A. 78: 149, 1999). Hemodialysis is now often done at home and this has brought the cost down to maybe $30,000/year; it's hard to find good figures.
Right now, we have 91,000 people in the US actually awaiting kidney transplantation, and 4100 die because they do not get a kidney (Lancet 379: 1461, 2012). If you start on hemodialysis and you do not get a transplant, you chance of being alive in five years is 35% (NEJM 357: 1316, 2007), and you will experience a great deal of malaise and fatigue.
All about dialysis: Lancet 353: 737, 1999. Today, a very sick patient with acute kidney failure may be placed on "continuous renal replacement therapy" instead of being dialyzed every few days; it's more expensive and it's not clear that outcomes are much better (Crit. Care Med. 31: 449, 2003; probably no better JAMA 299: 739, 2008).
* Ethics: Am. J. Kid. Dis. 15: 218, 1990 (classic article). What to do when somebody wants to discontinue and die: Am. J. Kid. Dis. 28: 147, 1996. The elderly seldom live long on dialysis: JAMA 271: 29 & 34, 1994. A search shows that there has been no "controversy" for the past decade about allowing folks to discontinue dialysis -- autonomy as a principle of medical ethics is once again triumphant.
You'll learn the criteria for classifying and staging "chronic kidney disease" from the clinicians (Lancet 379: 165, 2012). Whether you get kidney disease is mostly dumb luck. But whether it progresses to end-stage kidney failure does depend largely on what gets done about it.
Once kidney disease gets underway, it is self-perpetuating, causing sclerosis of the glomeruli. Inhibiting the renin-angiotensin system and (maybe) dietary protein restriction greatly slow the process. See Lancet 338: 419 & 423, 1991 (protein restriction is now challenged: Nat. Clin. Pr. Nephro. 3: 383, 2007). Update on the molecular biology ("Hold tight or you'll fall off", concerning podocytes J. Clin. Inv. 122: 13, 2012). Formulas for predicting the progression of severe disease to the end-stage: JAMA 305: 1553, 2011. Update on slowing the progression of renal disease (in-the-works are such things as endothelin blockers, anti-oxidants, antifibrotics, cytokines, statins-for-all, etc.): Am. J. Med. 118: 1323, 2005.
The JAMA proclaimed in 2007 that the prevalence of chronic kidney disease (i.e., microalbuminemia and elevated serum creatinine) in the US is increasing (1988-1994 vs. 1999-2004). However, most of this is due to more hypertension and diabetes in a fatter, longer-living population, and no one questions that the likelihood of progresion to dialysis dependence is less today.
This has come to a head. Nephrologists in Alberta are reporting that 70% of people referred to them for lab-detected "chronic renal failure" have nothing wrong with their kidneys (JAMA 303: 1151 & 1201, 2010).
* Historically, African-Americans are more at risk for end-stage kidney disease, for reasons that include the greater severity and frequency of hypertensive kidney disease in this population. Whether their problems are neglected by doctors, or treated less skillfully, or whether the real explanation lies elsewhere, seems to me to remain unanswered. The discrepancies are disappearing (or have perhaps completely disappeared when one controls for poverty: Med. Clin. N.A. 89: 419, 2005).
The Zuni people of our southwest have a terrible problem with both diabetic kidney disease and glomerulonephritis (Arch. Path. Lab. Med. 113: 148, 1989; ongoing Am. J. Kid. Dis. 39: 358, 2002; Am. J. Kid. Dis. 41: 1195, 2003; J. Am. Soc. Neph. 14(7S2): S-139, 2003). I would not be surprised if the cause is virus (betting on an undiscovered hantavirus), but this is not proved. By contrast, the pathologists at the New Mexico medical school think it is IgA nephropathy secondary to alcoholic liver disease (J. Histochem. Cytochem. 38: 699, 1990). Since the Zuni are highly inbred and genetics obviously plays a role in their IgA nephropathy (Kid. Int. 57: 1818, 2000; Am. J. Kid. Dis. 56: 251 & 289, 2010) as well, very likely we are looking at "a series of unfortunate events".
|
|
|
|
* In just one town, at least 66 people who wrecked their own kidneys by shooting heroin ("heroin nephropathy") were being maintained in the 1980's at a yearly cost of over $1.3 million to the public (JAMA 250: 2935, 1983). I doubt that things are much different today. The true prevalence and cost of heroin nephropathy are probably many times higher.
In the United States, society now balks at expensive, life-saving care for children who are in the country illegally. Dialysis, along with chemotherapy and transplantation, features prominently in these discussions (Pediatrics 114: 1316, 2004).
Foregoing dialysis for the severely mentally handicapped seems to be much more acceptable nowadays than in the past (Am. J. Med. Sci. 320: 374, 2000 -- "paternalism" is of course still wicked but "autonomy is out of the question").
Kidney failure due to acute tubular necrosis (nowadays, "acute renal injury", but the pathology is the same) is a common, potentially lethal complication in the intensive-care unit. Renal insufficiency due to underperfusion (dehydration, shock or a failing heart) or due to obstruction are extremely common.
High blood pressure commonly results from kidney problems, and in turn always damages the kidneys to some extent.
At least 10% of women will get acute pyelonephritis during their reproductive years, often during pregnancy.
At least 1% of people will pass a kidney stone (some give a number as high as 5%), producing some of the most severe pains in disease. Because of dehydration (hot temperatures, poor-quality drinking water), one out of every eight of our soldiers in Vietnam passed a kidney stone while there.
Kidney transplantation allows a near-normal, healthy life -- as long as the graft lasts. The histopathology is reviewed in Am. J. Surg. Path. 8: 243, 1984 (still good); NEJM 349: 2326, 2003 (how the changes progress with today's therapy); Curr. Op. Nephro. 193: 260, 2010 (how things work nowadays). See below.
I'll list the pathology of renal transplants at the end of the "kidney pathology" unit; we'll look closer in a later unit as it's quite difficult. Pathologists grade it by the 1995 Banff system. Since the worst problem is really vascular narrowing (arteries plus arterioles plus glomeruli), its severity can be estimated by measuring blood flow resistance using Doppler (NEJM 349: 115, 2003), perhaps sparing your patient a biopsy.
* Today's high-tech pathologists may offer a microarray assay on biopsy tissue to assess ongoing kidney cell injury. This is a strong prognosticator (J. Clin. Inv. 120: 1852, 2010).
The classic idea that the heart must still be beating if the kidney taken for transplant is to work well is now challenged by recent data (Br. J. Surg. 92: 113, 2005); this is holding up though grafts clearly do better if the kidneys are taken before cardiac death (J. Am. Coll. Surg. 212: 440, 2011).
One big surprise is that with today's immunosuppressive regimens, a kidney from your husband or wife lasts almost as well as "somebody who is a wonderful HLA match" (NEJM 333: 333 & 379, 1995). This discovery has transformed the whole business of transplantation, and has been a major boost to the practice, now common in some nations, of the living selling their organs to those in need. The risk of a living donor ending up with end-stage renal disease is only slightly increased: JAMA 311: 579, 2014. See below.
The glomerular diseases that tend to recur in transplanted kidneys are FSGS, membranous GN, membranoproliferative GN, and IgA nephropathy. Still, fewer than 10% of people with glomerulonephritis lose the graft to a recurrence (NEJM 347: 103, 2002.)
Worth knowing in the transplant era: Lymphocytes under the endothelium? "Endothelialitis" -- T-cell mediated transplant rejection!
Even more worth knowing: Grafts usually fail due either to antibody rejection or recurrence of the original glomerular disease.
|
|
REVIEW OF NORMAL ANATOMY AND PHYSIOLOGY
![]() Glomeruli forming
Glomeruli forming
in newborn
ERF/KCUMB
* According to most contemporary biologists, kidney design makes the most sense in light of its former functions in prehistoric life and present-day ocean creatures. Otherwise, the kidney is hard to understand.
Each adult kidney weighs around 150 grams and is composed of about 1 million nephrons that drain into about 14 calyces.
By forming urine, the kidney performs three important functions.
1. The kidney excretes the waste products of metabolism
A patient with any sort of impaired kidney function will have increased creatinine and urea nitrogen in the blood, or azotemia. If the kidney is adequately perfused, itself normal, and its outflow not obstructed, blood urea nitrogen levels will remain within normal limits.
2. The kidney regulates the body's content of water, sodium, potassium and calcium, and gets rid of excess phosphate.
|
| Hypertension, edema, and/or hyperkalemia may develop in renal disease. Renal edema is first visible around the patient's eyes. |
3. The kidney maintains the appropriate acid-base balance of plasma
Metabolic acidosis is characteristic of severe renal failure, because you cannot get rid of the sulfate and phosphate ions that you generate from burning your food.
The kidney also makes renin (REE-nin, please) and erythropoietin, and activates vitamin D.
High blood pressure, anemia, and bone demineralization are common in serious kidney disease.
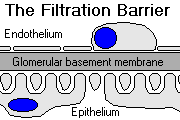
Endothelial cells have fenestrae (70-100 nm in diameter) that give plasma free access to the glomerular basement membrane.
Glomerular basement membrane ("GBM") is largely special collagen (type IV) that is the major permeability barrier. It also contains polyanions (heparan sulfate, perlecan, etc.) and other substances that are additional barriers.
* The alleged division of the GBM into lamina rara interna, lamina densa, and lamina rara externa is an artifact of no consequence. Ignore it in "Big Robbins".
Visceral epithelial cells ("podocytes") display interdigitating cell processes (foot processes) that grasp the capillaries. The foot processes are separated by "filtration slits" that are bridged by diaphragms with little rectangular pores (4 x 14 nm).
{46463} scanning electron micrograph of podocytes
The whole filter (essentially the GBM) is very permeable to water and small solutes, but it excludes most albumin and larger proteins from the filtrate.
The permeability barrier is size-dependent (3.5 nm), and is also charge-dependent (the polyanions exclude albumin and other anionic macromolecules.)
Loss of polyanions will let albumin through (selective proteinuria). If the filtration barrier is severely damaged, larger proteins will leak out (nonselective proteinuria). A leaky GBM results in the nephrotic syndrome.
If some of the capillaries rupture, there will be hematuria. If the capillaries are badly enough damaged to release much fibrin, the glomerulus will probably get replaced by scar tissue, and then the underperfused tubule will atrophy and probably disappear.
Glomerular structure and function:
The glomerulus is essentially a tuft of around 50 capillaries, each of which is a unit of the filter. They are surrounded by Bowman's capsule, which encloses the urinary space.
The capillaries in a glomerulus arise from one afferent arteriole and drain into one efferent arteriole.
They tend to group loosely into lobules of about ten capillaries each. Don't expect to be able to distinguish the lobules of a healthy glomerulus.
Glomerular capillary pericytes are called mesangial cells.
These produce mesangial matrix, which is the supporting framework for the GBM and is chemically the same.
Mesangial cells are contractile, helping regulate flow and filtration rate in the glomerular tuft (Am. J. Kid. Dis. 16: S-2, 1990) and probably phagocytize most things that shouldn't have gotten through the filtration membrane.
* Tip: In looking at an electron micrograph of glomerulus, orient yourself by finding something you are sure is a red cell, or are sure is the GBM.
The parietal epithelial cells that surround the tuft, together with their basement membrane, make up Bowman's capsule.
As noted, the capsule encloses the "urinary space" and is continuous with the proximal convoluted tubule.
Glomerular filtration rate ("GFR", i.e., the volume of plasma filtered into the urinary space per minute) should be about 120 mL/min for an adult. (GFR is estimated clinically by measuring creatinine clearance.)
Naturally, this varies directly with blood pressure, which in turn reflects the volume of fluid that is effectively circulating.
A working definition of adult chronic kidney disease is a real GFR<=60 for three months, or albuminuria >=30 mg/gm of creatinine. You may learn other classifications of renal severity, but always your clinical judgement will be paramount.
The juxtaglomerular apparatus is a group of special cells at the poles of nephrons formed from both afferent arteriole and distal tubule.
They are sensitive to volume, pressure, and sodium concentration in both.
They produce renin and adjust the GFR (by constricting the afferent arteriole, etc.) as necessary to maintain adequate systemic blood pressure.
Renin generates angiotensin II, which in turn raises systemic blood pressure by constricting arterioles, causing thirst, and causing production of aldosterone.
Renin isn't produced when microvascular disease has damaged the JGA. This is probably an important mechanism of "renal tubular acidosis type 4", a popular diagnosis, in which there is underproduction and/or under-response to aldosterone (hence, often hyperkalemia).
Tubular function:
The PROXIMAL CONVOLUTED TUBULE reabsorbs substances from the glomerular filtrate, including ions, glucose, phosphate, bicarbonate, amino acids, vitamins, and the smallest proteins (albumin, beta2-microglobulin), in isotonic solution. Of course, most of the sodium, potassium, chloride, and water in the glomerular filtrate are absorbed here, too.
The proximal convoluted tubule also secretes para-aminohippuric acid, uric acid, etc., and probably makes erythropoietin.
A patient with impaired function of the proximal convoluted tubule will lose substances in the urine (glycosuria, amino-aciduria, microglobulinuria, potassium, "renal tubular acidosis type 2").
The DISTAL NEPHRON retains or excretes water, ions, and protons, as required for homeostasis.
A patient with impaired function of the distal nephron cannot concentrate urine (hyposthenuria / nephrogenic diabetes insipidus).
The patient will first notice this when he or she starts having to get up at night to urinate (nocturia).
In severe disease, the specific gravity becomes fixed at 1.010 (iso-osmotic urine; "isosthenuria" -- healthy folks can usually dilute to 1.003 and concentrate to 1.030).
A patient with impaired function of the distal nephron often cannot dispose of protons (renal tubular acidosis type 1; these patients also tend to become hypokalemic -- why?)
Some genetic forms involving ion exchangers are known, but most result from renal or systemic disease.
* Untreated, the most severe cases can result in "Tiny Tim's disease" (Arch. J. Dis. Child. 146: 1403, 1992), with growth retardation, crippling and malforming osteomalacia (failure of bone to mineralize), and episodes of weakness.
"Renal tubular acidosis type 3" is just a combination of types 1 and 2.
The LOOP OF HENLE is responsible for maintaining a hypertonic interstitium in the medulla. This is the famous "countercurrent multiplier" mechanism.
The DISTAL CONVOLUTED TUBULE is the site of sodium (and hence water) resorption, and of potassium and proton ("fixed acid") excretion.
A high GFR produces rapid flow of filtrate through the distal convoluted tubule, resulting in little sodium resorption. A low GFR will have the opposite effect. This of course is important in regulating intravascular fluid volume and blood pressure.
The distal convoluted tubule is also influenced by aldosterone, which promotes sodium retention and potassium and proton loss. Aldosterone comes from adrenal gland under stimulus of renin-angiotensin and of salt-and-water depletion.
The COLLECTING DUCT is site of anti-diuretic hormone (hADH) action.
This neuropeptide is produced when osmoreceptors in the hypothalamus determine the need for the body to retain water. It opens little pores in the walls of the collecting ducts, allowing water to flow back into the hypertonic renal interstitium.
Inability of the collecting duct to respond to hADH produces nephrogenic diabetes insipidus.
"Atrial natriuretic factor" (hANF, atriopeptins, etc.), the most important of several natriuretic peptides (NEJM 399: 321, 1998). It comes from the atria, cause loss of water and sodium by several mechanisms. It's released when the right atrium is stretched. This is probably the overriding way in which we regulate our extracellular fluid volume in health.
ANF...
Tubular diseases that prevent reabsorption of water (or a non-resorbable substance in the filtrate) will produce polyuria (urine volume more than 1500 mL/day). Plugged or leaky tubules (or low GFR) will cause oliguria (urine volume less than 500 mL/day.)
Casts in the urinary sediment are cylinders of congealed Tamm-Horsfall protein produced by the tubular cells. They may contain other formed elements that aid in the diagnosis of kidney disease.
Hyaline casts do not contain formed elements, and are a normal finding.
Epithelial casts contain renal tubular cells and suggest interstitial disease or acute tubular damage. Fatty casts are epithelial casts in which the cells contain abundant lipid (i.e., the patient has the nephrotic syndrome.)
Red cell casts ("active sediment") indicate bleeding into the nephron (i.e., glomerular disease). Hemoglobin casts usually mean the red cells have hemolyzed, often in the bloodstream.
{17244} red cell cast
White cell casts contain polys and indicate acute inflammation in the renal interstitium.
Granular casts are cellular casts in which the cells have undergone necrosis and fragmentation.
Casts that contain a lot of lipid mean nephrotic syndrome (which you should already be aware is present.)
Broad and waxy casts are very large casts that indicate a low rate of flow through the tubules and hence serious disease.
Renal interstitium:
The interstitium in the cortex is scanty, but in the medulla it is responsible for maintaining the ability of the urine to be concentrated.
Damage to the interstitium will result in inability to concentrate the urine.
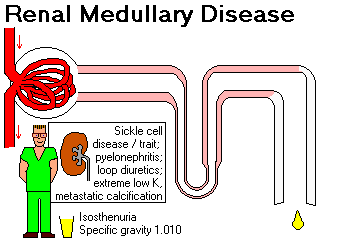
Vessels:
Blood to the kidney goes successively through the renal artery, its primary divisions, the interlobar arteries, the arcuate arteries, the interlobular arteries, the afferent arterioles, the glomerular capillaries, the efferent arterioles, the intertubular capillaries (including vasa recta), and finally into the veins.
All the blood that supplies one nephron flows through the glomerulus first. If the glomerulus dies, the whole nephron dies.
Narrowing of the arteries and/or arterioles supplying some or all of the kidney tissue will produce systemic high blood pressure.
Blood pressure in the glomerulus is lower than it should be, resulting in too little filtrate being produced, and too much sodium and water being resorbed in distal convoluted tubule.
Plus, the juxtaglomerular apparatus produces too much renin. Most high blood pressure resulting from kidney disease is "high renin" hypertension.
SYNDROMES OF KIDNEY DISEASE: Am. J. Kid. Dis. 10: 181, 1987 (still good)
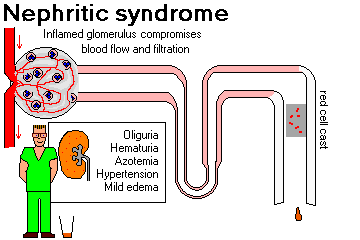
NEPHRITIC SYNDROME ("nephritis")
Indicates acute inflammation of glomeruli
If the process continues, the glomerulus may be destroyed (i.e., rapidly progressive glomerulonephritis or chronic glomerulonephritis will result.)
You will want to remember that each of these is likely to cause the nephritic syndrome:
Note that all of these except anti-GBM disease, Wegener's, and polyarteritis are immune-complex diseases.
NEPHROTIC SYNDROME ("nephrosis", an archaic term): NEJM 338: 1202, 1998 (great photos); kids Lancet 362: 629, 2003; BMJ 336: 1185, 2009 for diagnosis and management.
Indicates excessive permeability of the filtration membrane to plasma proteins.
In mild cases, only the small plasma proteins (albumin, etc.) will be lost, and the patient has selective proteinuria. Severe cases show nonselective proteinuria.
Some cases of the nephrotic syndrome may resolve with or without treatment. Longstanding heavy proteinuria is itself harmful to the kidney and will lead to renal failure after several years.
Although the patient is edematous and has increased total body water, the lack of plasma protein results in a loss of effective circulating volume. This in turn causes salt and water retention and accumulation of yet more fluid in tissue spaces. Secondary hyperaldosteronism also plays a part in causing the edema.
NEPHRIN ("heparan binder") is a protein normally located along the surfaces of podocytes, with its pairs forming the basic structure of the "slit diaphragm". Several different molecules involved in immune injury can cause it to be lost from the cell surfaces, either by allowing the polyanions to escape, or from the slit diaphragms being lost. In each case, the nephrotic syndrome results (Am. J. Path. 158: 1723, 2001), and this is probably the common denominator for most (maybe all) causes of nephrotic syndrome.
Hyperlipidemia is due, at least in part, to increased production of lipoproteins by the liver to compensate for the loss of albumin. This is a modest coronary risk, and you may treat it with statins (Am. J. Card. 76: 97A, 1995.)
Patients with nephrotic syndrome are at increased risk for thrombosis throughout the body. Various explanations have been proposed; easiest to believe is loss of antithrombin 3 in the urine.
![]() Renal vein thrombus
Renal vein thrombus
WebPath Photo
And patients with nephrotic syndrome are generally at increased risk for thrombosis. There are numerous explanations given; perhaps the easiest to understand is heavy urinary loss of antithrombin 3.
Nephrotic syndrome patients are also very prone to infection, notably with gram-positive cocci ("cellulitis", "primary pneumococcal peritonitis", etc., etc.) Loss of complement factors B and D is cited as a cause, and of course there is also iatrogenic immunosuppression.
Finally, the nephrotic kidney is extra-prone to sudden shutdown (probably a combination of prerenal azotemia and acute tubular necrosis; see Am. J. Kid. Dis. 19: 201, 1992).
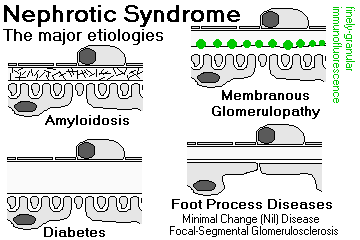
Causes of the nephrotic syndrome:
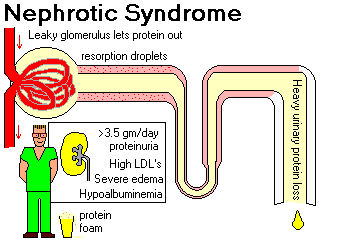
{16764} nephrotic syndrome
{16857} kidney, yellow cortex of nephrotic syndrome
{16800} lipid in tubule, nephrotic syndrome, oil red O
(lipid is red)
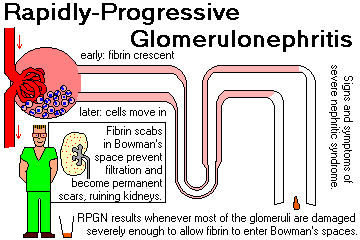
RAPIDLY PROGRESSIVE GLOMERULONEPHRITIS ("RPGN")
Indicates sudden, severe injury to most of the glomeruli.
The nephritic syndrome will probably also be present.
The common denominator is that the glomerular basement membrane is ruptured, and fibrin (and hence "crescents" -- unwholesome things made of fibrin and mixed cells) is present in Bowman's space.
You'll want to remember these as causes...
The three categories (I, II, and III) are about equally common. Note that this list overlaps with nephritic syndrome, which is how most RPGN starts out.
ASYMPTOMATIC HEMATURIA (gross or microscopic):
Mild nephritic-type diseases may produce only microscopic hematuria. Of course, there is always proteinuria when there is hematuria.
Red cell casts are proof that hematuria originates in the nephron (i.e., the patient probably has glomerular disease).
Kidney stones, sickle cell nephropathy, bleeding disorders, and cancers are the other important diseases that produce bleeding at the level of the kidney. These seldom produce red cell casts.
IgA nephropathy is also likely to fall into this category.
HEMOLYTIC-UREMIC SYNDROME
Endothelial damage and fibrin-rich microthrombus formation in the renal vascular bed, resulting in evidence of red cell fragmentation (schistocytes) and renal failure.
There are many different causes, including an infectious disease, mostly affecting young children caused by vicious E. coli, shigellosis, estrogen effects (oral contraceptive, pregnancy), malignant high blood pressure, vasculitis, etc., etc.
PYELONEPHRITIS
Bacterial infection of the kidney generally produces fever, flank pain, proteinuria, pyuria (PMN's in the urine), and white cell casts.
PROXIMAL TUBULE DYSFUNCTION
A group of conditions (mostly not-so-serious) in which the proximal tubule fails to conserve one or more substances, which are lost in the urine.
In mild impairment, small-molecular weight proteins (beta2- microglobulin, light chains, * lysozyme, * retinol-binding protein, * vitamin-D binding protein) are lost in the urine (tubular proteinuria).
When the proximal tubule is seriously impaired (the various renal Fanconi syndromes), there is wasting of bicarbonate ("renal tubular acidosis type 2"), glucose ("renal glycosuria"), calcium (kidney stones, rickets, osteomalacia), phosphate, potassium, amino acids, other small molecules, etc. etc.

Kidney failure: loss of renal function.
ACUTE RENAL FAILURE usually presents as oliguria (less than 500 mL urine/day) plus azotemia.
Hyperkalemia is a great threat to life during the oliguric phase. Be vigilant, and keep it from killing your patient.
Remember that medicines that are eliminated by the kidney must be curtailed. Whether or not "medicines are the fourth-commonest cause of death in the USA", plenty of patients die because someone doesn't realize somebody is in renal failure.
CHRONIC RENAL FAILURE is the end result of irreversible kidney damage from any cause.
Sometimes the cause of the "end-stage contracted kidney" cannot be determined even at autopsy. Once a sufficient number of nephrons are destroyed, the rest of them start dying off too.
Signs and symptoms are those of uremia.
Anuria, the complete cessation of urine production, is rare in acute renal failure -- the major exception is diffuse cortical necrosis. It may occur late in chronic renal failure. Much more often, anuria is due to obstruction of ureters or urethra or (most often) Foley catheter. (Some people will define "anuria" to be "less than 100 mL/day").
* Spontaneous recovery of renal function is rare but does occur (around 1%, more in patients with lupus; see Am. J. Kid. Dis. 15: 61, 1990.)
HIGH BLOOD PRESSURE ("hypertension"): In kidney failure from any cause, blood pressure will rise for various reasons (Am. J. Kid. Dis. 32: 705, 1998).
"Renal" hypertension results from decreased GFR and/or increased renin (usually stenosis of arteries).
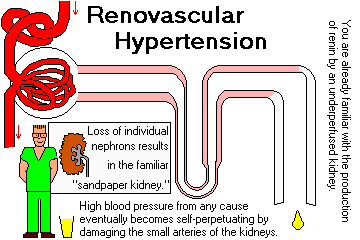
In time, the high blood pressure will surely cause further damage to the kidney arteries, producing a vicious cycle (Am. J. Kid. Dis. 19: 484, 1992 is stil good).
KIDNEY STONES: Flank pain and hematuria (but no red cell casts). Secondary infection is common.
PROGRESSIVE LOSS OF REMAINING RENAL FUNCTION: Once the kidney is damaged to a certain degree, it continues to deteriorate (i.e., undergo more scarring, notably glomerular sclerosis) even if the underlying disease is cured.
How this happens is still somewhat mysterious. In health, mesangial matrix is normally synthesized and degraded over time. In progressive renal failure, something slows the degradation.
Contributing factors include hypertension, hyperlipidemia, high dietary protein, and high dietary phosphate.
* Many molecules are involved; in particular, excess plasminogen activator inhibitor-1 seems to prevent the normal breakdown/recycling of matrix by t-PA (review J. Clin. Inv. 112: 379, 2003), and this is a possible target for therapy (J. Clin. Inv. 112: 326, 2003).
* The new player is the Notch pathway, which we might target with a gamma-secretase inhibitor. Well, it works in rats.... Nat. Med. 14: 290, 2008.
* Apolipoprotein E genotype influences the rate of progression, with episilon4, which is bad elsewhere, being good here (JAMA 293: 2892, 2005.)
This part of the process can be slowed with anti-hypertensive therapy (especially angiotensin converting enzyme inhibitors; see Am. J. Kid. Dis. 31: 161, 1998 and NEJM 334: 939, 1996 were the great historic articles) and dietary protein restriction.
And it turns out that angiotensin II actually mediates a lot of the fibrosis in longstanding renal disease. When its receptors are stimulated, there's local production of a variety of substances (notably TGF-beta1, TNF-alpha, PDGF A-chain) that are implicated in the deadly scarring-up of glomeruli and vessels (Am. J. Kid. Dis. 31: 171, 1998.)
Another "usual suspect" is indoxyl sulfate, a breakdown product from the diet (J. Lab. Clin. Med. 124: 96, 1994; Am. J. Kid. Dis. 37 (1S2): S7-12), 2001).
And nowadays we are using statins to slow this process when the underlying cause is nephrotic syndrome (Am. J. Kid. Dis. 15: 16, 1990, Clin. Pharm. Ther. 67: 427, 2000, lots of others).
UREMIA: The clinical signs and symptoms of renal failure; especially, the clinical problems associated with chronic renal failure. Today, the definition excludes electrolyte, calcium, blood pressure, and vitamin D problems. Update NEJM 357: 1316, 2007.
AZOTEMIA means increased urea and/or creatinine in blood from any cause.
When kidney function falls below about 10% of normal, uremia becomes apparent.
Fluid, electrolyte, and acid-base disturbances:
Calcium - phosphorus problems
Bone problems: the miserable "renal osteodystrophy" that includes:
No one really knows why the parathyroid glands undergo hyperplasia and become overactive in renal failure. The obvious explanation (lack of vitamin D along with too much blood phosphate) is supported by the finding that simply giving oral vitamin D helps (update J. Clin. Endo. Metab. 91: 2480, 2006; even a short high-dose course works marvels Am. J. Clin. Nutr. 95: 522, 2012).
You also want to be sure you know what's going on... if the parathyroids are large and the main problem is secondary hyperparthyroidism (bloodwork, perhaps bone biopsy), the parathyroids can be trimmed surgically; if the problem is one of the other two illnesses ("quiet bone"), parathyoridectomy is contra-indicated. I hope this makes sense.
Uremic toxins causing altered gene expression may be part of the picture as well (J. Clin. Endo. Metab. 91: 563, 2006).
Exactly what causes the osteomalacia isn't fully understood, either.
* A new player is fibroblast growth factor 23, known to us from paraneoplastic phosphate wasting. When elevated early in the course of kidney disease, it predicts a poor outcome (JAMA 305: 2432, 2011).
Cardiopulmonary:
* The most likely explanation is that something toxic (very likely, advanced glycosylation end-products) that are ordinarily filtered by the kidney are not being removed by dialysis (Lancet 343: 1519, 1994).
Hematopoietic:
Recombinant human erythropoietin (* "Epogen") has been a big help.
* It costs perhaps $6000/patient per year, yet was originally spawned by the orphan drug act of 1983.
![]() Cardiorenal Anemia Syndrome Academy
Cardiorenal Anemia Syndrome Academy
Friends of mine who
examine novel therapies
GI:
Skin:
One cause of pruritus is calcium sulfate and calcium phosphate precipitating in the sweat. Others include excesses of vitamin A metabolites and histamine.
* "Calcinosis cutis" affects about 1% of dialysis patients; it's a calcification syndrome caused by elevated serum calcium (Arch. Derm. 142: 900, 2006) that expresses itself only if, for some reason, osteopontin is locally produced. Picture NEJM 357: 2615, 2007.
{24577} uremic frost
Neuromuscular:
The old "dialysis dementia" was due to aluminum accumulation and is still studied. Aluminum seems to tangle tau protein in neurons, forming the same paired helical filaments seen in Alzheimer's: Lancet 343: 993, 1994.
It's claimed that mentation slows when GFR drops below 50% of normal (J.Am. Soc. Neph. 18: 2205, 2007).
* Only during the 1990's (perhaps coinciding with the advent of managed care), did people being writing about what to do to make the death of a person refusing dialysis more pleasant (Lancet 346: 3 & 506, 1995). The death can be "good" if the "war on drugs" doesn't prevent the patient from receiving pain medicine (Arch. Int. Med. 155: 42, 1995), etc., etc.
Infections:
* One factor is loss of Fc receptors on macrophages: NEJM 332: 717, 1990.
Other:
{05904} peritoneal dialysis guy
{05905} peritoneal dialysis, how-to
{05913} hemodialysis, fistula
{05914} hemodialysis machine
KIDNEY BIOPSY (standards J. Clin. Path. 49: 233, 1996; J. Clin. Path. 53: 433, 2000)
For a better understanding of what is going on in your patient's kidneys, you (or, better, the nephrology consultant) can obtain a piece of one for the pathologist using a special hollow "needle" that is passed in through the skin.
You will learn the indications for this procedure on rotations. Nowadays, kidney biopsies can be obtained by your radiologist via the transjugular approach (Radiology 215: 689, 2000).
Remember: Hyaline casts in the urine are normal. Other casts in the urine indicate disease in the nephron.
INTRODUCTION TO GLOMERULAR DISEASE (update for clinicians Arch. Int. Med. 161: 25, 2001; pathologists Med. Clin. N.A. 81: 653, 1997)
Dr. Richard Bright of Guy's
Had several patients large in size.
Their legs were swollen as could be;
Their eyes so puffed they could not see.
To this edema Bright objected,
And so he had them venesected.
He took a teaspoon by the handle,
Held it over a tallow candle,
And boiled some urine over the flame,
(As you or I might do the same.)
To his surprise, we find it stated,
The urine was coagulated.
Alas, his dropsied patients died.
Our thoughtful doctor looked inside:
He found their kidneys large and white,
Their capsules were adherent quite.
So that is why the name of Bright is
Associated with nephritis.
-- Anonymous!
This is the most difficult lecture in Medical Pathology.
Glomerular diseases are classified according to
To make matters worse, the mechanisms of glomerular injury are often obscure. It is difficult or impossible to correlate abnormal structure and abnormal function for most glomerular diseases.
Some definitions:
DIFFUSE (all glomeruli) vs. FOCAL (only some glomeruli, maybe under 80%)
GLOBAL (entire glomerulus) vs. SEGMENTAL (a part of a glomerulus)
Global diseases are usually diffuse, and segmental diseases are usually focal. The one major exception is glomerular damage from high blood pressure, which is focal-global fibrosis. (Why? Hint: Each glom has exactly one afferent arteriole.) Unless otherwise specified, the diseases we will describe in this lecture are global-diffuse.
* HYALINOSIS ("fibrinoid"): deposits of plasma proteins. (This stuff doesn't stain blue with "trichrome" or black with "silver", distinguishing it from fibrosis and sclerosis respectively.)
SCLEROSIS: enough increase in basement membrane - mesangial matrix material to compromise the lumens of capillaries. (The distinguishing feature is that this stains positive with silver).
FIBROSIS: type I collagen, i.e., an organized scar. (Blue on "trichrome". Unlike hyalinosis and sclerosis, this is essentially PAS-negative.)
HISTOLOGIC ALTERATIONS IN GLOMERULAR DISEASE
Endothelial and mesangial cells may proliferate (intracapillary proliferation).
This tends to narrow and occlude the capillaries.
Why these cells proliferate in some disease states is generally unknown.
Mesangial cell proliferation, so common in glomerular disease, seems to have as a major cause platelet-derived growth factor, and the mesangial cells themselves seem to produce a protein that turns this potent stimulus off as recovery begins. Am. J. Path. 148: 1153, 1996. In the meantime, several drugs are under investigation to inhibit this proliferation (Kid. Int. 68: 474, 2005, others).
Visceral and parietal epithelial cells and fibroblasts may also proliferate (extracapillary proliferation).
Proliferation of the visceral and parietal epithelium is seldom striking, and you'll want special stains to appreciate it.
If the fibroblasts proliferate, it's probably because of fibrin leaking out of glomerular capillaries.
If the fibrin leak is small, a fibrous adhesion between a few capillaries and Bowman's capsule may result.
If the fibrin leak is big, a cellular "crescent" soon fills Bowman's space, which ultimately becomes a crescent-shaped fibrous scar.
Crescents indicate the glomerular basement membrane itself has been seriously damaged. To make things worse, the crescent will both squish the glomerular capillaries and also obstruct the outlet to the proximal tubule.
Polymorphonuclear leukocytes in the glomerulus indicate complement is being fixed there. Enzymes from polys (as well as complement itself) will damage the glomerulus.
Polys almost never cross the GBM. Regardless of how severe the acute inflammation is in the glomerulus, the polys will not appear in the urine.
* You'll need special stains to see if monocyte/macrophages are present.
This is highly characteristic of several common causes of the nephrotic syndrome (and is the characteristic finding in minimal change glomerulopathy and focal-segmental glomerulosclerosis). Injured epithelial cells swell, and this obliterates the discrete foot processes.
This is almost always accompanied by focal detachment of the cells from the GBM. Of course, the underlying problem is disruption of the cytoskeleton as all this happens: Am. J. Path. 148: 1283, 1996. The molecular biology is now being worked out (* "dynamin" is a molecule responsible for maintaining podocyte actin structure, and it is cleaved by a cathepsin in these diseases: J. Clin. Inv. 117: 2095, 2007.)
The GBM may actually be thicker (as in diabetic glomerulosclerosis and membranoproliferative glomerulonephritis.)
The GBM may appear thicker because of immune-complex deposits ("electron-dense deposits", etc.) These are just barely visible by light microscopy.
They may be subendothelial, intramembranous, subepithelial or mesangial in location. (Why immune complexes localize where they do is still mysterious.)
Hyaline material is deposited subendothelially in certain capillary loops. The stuff stains dark pink and is lipid-rich. It includes the old "fibrin caps" of diabetes, and the changes seen in solitary kidneys (congenital, following massive infarcts or subtotal nephrectomy, etc) and other over-perfused kidneys -- even live kidney donors eventually get hyalinosis, proteinuria, and occasionally impaired renal function (J. Urol. 152: 312, 1994 -- the upshot, though, is that most live kidney donors have a greatly improved quality of life because of their generous act JAMA 305: 592, 2011).
The common denominator seems to be hyperfiltration.
This is seen in diabetes (nodular glomerulosclerosis or Kimmelstiel-Wilson disease) and light-chain disease, and occasionally in malignant hypertension, glomerulonephritis, DIC, HUS, radiation injury, graft-vs.-host, chronic antibody-mediated transplant rejection, sicklers (Am. J. Kid. Dis. 15: 361, 1990), and cobra envenomation.
This is seen in the most severe glomerular disease. Karyorrhexis of glomerular cells (not just PMN's) is the surest sign of necrosis. (Look for "fibrinoid", too, of course. Later, there is obvious red infarction.)
More insidious processes show no necrosis on renal biopsy, but destroy the kidney just as effectively.
Early ischemic changes in the glomerulus include corrugation and irregular thickening of the GBM and Bowman's basement membrane. It's as if it crumples as it deflates.
Later, the whole glomerulus is replaced with collagen ("obsolescent"). The tuft can usually be identified as a PAS-positive nubbin at the vascular pole.
* JGA hyperplasia is seen in chronic ischemia of the glomerulus, but there are better ways to detect renal vascular disease....
* The molecular mechanisms that produce fibrosis in and around the ischemic glomerulus are still poorly-understood (Am. J. Path. 176: 594, 2010).
This is evidence of chronic, irreversible damage.
Hyalinization of the tuft itself may be:
Hyalinization around the tuft (i.e., in Bowman's space) is usually type I collagen. It may be:
* Do not confuse either of these with "hyalinosis" lesion, which looks like lipstick smudges in serious glomerular disease.
MECHANISMS OF GLOMERULAR INJURY
Most of the known mechanisms involve antibody-antigen complexes. These include:
In situ antibody deposition / immune complex formation
Anti-GBM antibodies (i.e., experimental Masugi nephritis, clinical Goodpasture's syndrome and its variants): discrete granular deposits are not seen, but linear deposition is seen on immunofluorescence, and eluates from diseased kidneys deposit in linear fashion on normal kidney.
Antibodies against other fixed antigens (i.e., experimental Heymann nephritis caused by antibodies against megalin in the proximal tubule brush border Am. J. Path. 146: 1481, 1995; evenly-spaced, fine granular deposits are seen on immunofluorescence. All about Heymann nephritis: Am. J. Path. 144: 807, 1994.)
Antibodies against planted antigens (* i.e., drugs that bind to the GBM)
Circulating immune complex deposition (* i.e., experimental and clinical serum sickness, systemic lupus, acute post-streptococcal glomerulonephritis): coarse granular deposits are usually seen on immunofluorescence.
* Cationic complexes tend to localize subepithelially or subendothelially; neutral and anionic complexes tend to localize in the mesangium. Remember IgA is Anionic. The most strongly anionic complexes don't make it through the GBM.
Immune-complex-related injury is mediated by complement activation, polys, perhaps also macrophages, coagulation system, etc. etc.
* One doesn't always know the significance of immune complexes. They may have formed elsewhere and ended up stuck in the kidney, or formed in situ following exposure of a sequestered antigen, or have accumulated in damaged tissue, or be of no importance at all. For example, the clumps of IgM-rich material that are seen in around 20% of transplanted kidneys at biopsy do not seem to mean much of anything (Arch. Path. Lab. Med. 129: 231, 2005).
* Review of immune-based kidney disease: J. Allerg. Clin. Imm. 111(2-S): S-637, 2003.
Much glomerular damage (hereditary nephritis, diabetic glomerulosclerosis, late changes of high blood pressure, etc.) is clearly not immune-mediated.
DIFFUSE PROLIFERATIVE GLOMERULONEPHRITIS
{13388} diffuse proliferative glomerulonephritis, fatal case, flea-bitten cortex
{16814} diffuse proliferative glomerulonephritis, H&E
{08261} diffuse proliferative glomerulonephritis
{08264} diffuse proliferative glomerulonephritis, low power
{09709} diffuse proliferative glomerulonephritis (this was a mild case of post-streptococcal disease)
{17020} diffuse proliferate glomerulonephritis (this was a case of post-streptococcal disease)
{24845} diffuse proliferative glomerulonephritis (this was a case of post-streptococcal disease)
{24846} diffuse proliferative glomerulonephritis (this was a case of post-streptococcal disease)
{24847} diffuse proliferative glomerulonephritis, immunofluorescence (this was a case of
post-streptococcal disease)
{17024} diffuse proliferative glomerulonephritis, immunofluorescence
{09718} diffuse proliferative glomerulonephritis, coarsely-granular immunofluorescence (this was a
case of post-streptococcal disease)
{09721} diffuse proliferative glomerulonephritis, electron micrograph (this was a case of
post-streptococcal disease)
{16769} diffuse proliferative glomerulonephritis, red cell cast
{08267} diffuse proliferative glomerulonephritis, coarsely-granular immunofluorescence pattern
{08270} diffuse proliferative glomerulonephritis, electron- micrograph showing large subepithelial
deposits
{16817} diffuse proliferative glomerulonephritis, EM
|
|
ACUTE POST-STREPTOCOCCAL![]() GLOMERULONEPHRITIS
(Am. Fam. Phys. 71: 1949, 2005)
is the prototype and commonest cause of this reaction pattern.
GLOMERULONEPHRITIS
(Am. Fam. Phys. 71: 1949, 2005)
is the prototype and commonest cause of this reaction pattern.
This produces the nephritic syndrome in kids two weeks or so following a respiratory or skin infection with a "nephritogenic strain" of group A, beta-hemolytic streptococci.
The cause is deposition of circulating immune complexes that fix complement and attract PMN's. THERE IS NOT, AND NEVER WAS, A STREPTOCOCCAL INFECTION INVOLVING ANY PORTION OF THE KIDNEY. (* Streptococcal antigens also activate properdin.)
* The immunology seems to involve a brisk response by the complement pathway to a portion of the M-protein of streptococcus (J. Immunol. 148: 3110, 1992; J. Ped. 131: 293, 1997.).
In the molecular mayhem that follows, the glomerular capillaries are damaged.
Some capillaries rupture, causing gross hematuria.
The endothelial cells in all the glomeruli swell up, proliferate, and choke off their blood supply, making the glomeruli hypercellular and bloodless. (This explains the oliguria, edema, and hypertension.)
* If many capillaries have been badly damaged (which is rare in children but fairly common in adults), there may also be extracapillary proliferation (crescent formation)
Polys are easy to find in the glomerular tufts, but do not cross the glomerular basement membrane into the urine. (This makes the glomeruli appear even more cellular.)
Immunofluorescence shows coarse granular deposits containing immunoglobulin and complement.
Electron microscopy shows these granules to be large, dense, hump-shaped deposits located subepithelially (i.e., on the epithelial side of the GBM).
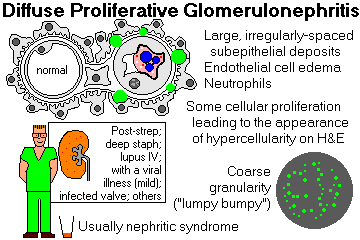
* Lab findings that support the diagnosis of a streptococcal etiology include increasing anti-streptolysin O ("ASO") titers and decreased C3 levels in the serum.
Kids are likely to recover, but an adult, especially a diabetic, is likely to have serious permanent damage (Medicine 87: 21, 2008).
Other causes of diffuse proliferative GN include:
* Post-viral glomerulonephritis is a trivial disease with microhematuria and depressed complement levels but no other symptoms or signs. You'll probably never make this diagnosis, but you may well have had this semi-disease yourself.
RAPIDLY PROGRESSIVE GLOMERULONEPHRITIS (RPGN, "crescentic glomerulonephritis"):
{08907} rapidly progressive glomerulonephritis, H&E
{16837} rapidly progressive glomerulonephritis, trichrome
{16838} rapidly progressive glomerulonephritis, IF for fibrin
{16839} rapidly progressive glomerulonephritis, PAS stain
{34228} rapidly-progressive glomerulonephritis with crescent
|
|
This syndrome involves rapid, loss of renal function (typically loss of at least 50% of renal reserve over 3 months), usually with the nephritic syndrome.
The morphologic correlate is severe glomerular injury (i.e., many crescents). There are several familiar causes, and you should know the contemporary classification:
RPGN I: anti-GBM disease
RPGN II: RPGN superimposed on any immune complex disease
RPGN III: RPGN without significant immune deposits; usually with systemic vasculitis syndromes
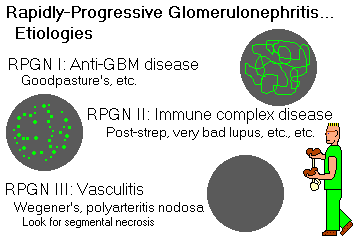
RPGN I: ANTI-GBM DISEASE (Goodpasture's, Masugi, etc.) (20% of RPGN cases)
{16842} Goodpasture's, IF
{16843} Goodpasture's, IF
{16847} Goodpasture's, IF
{16851} Goodpasture's, silver stain
{17073} Goodpasture's, IF
{17074} Goodpasture's, IF for fibrin
{34234} Goodpasture's, IF
The patient makes antibodies against an antigen uniformly distributed along the GBM. The autoantigen is a portion of a chain of type IV collagen. Update on the antigen: NEJM 363: 343 & 388, 2010).
GOODPASTURE'S DISEASE (South. Med. J. 95: 1411, 2002) / ANTI-GBM DISEASE. Goodpasture's is anti-GBM disease with RPGN and lung hemorrhages. (The auto-antibody cross-reacts with the pulmonary basement membrane.) It usually occurs in young men. Patients commonly asphyxiate on their own blood after a few months, unless they die of renal failure first.
The cause of Goodpasture's syndrome remains unknown. A link to hydrocarbon exposure is classic; unfortunately, some of the epidemiologic studies have been ruined by fictitious data (Acta. Med. Scand. 218: 256, 1985.) One known cause is the drug penicillamine, which is thought to expose the antigen by breaking sulfhydryl linkages.
More generally, the immune response seems to follow some unexplained alteration in the tertiary structure of the antigen itself ("conformeropathy"): NEJM 363: 343, 2010.
Plasmapheresis and immunosuppression remain effective treatments for anti-GBM disease, and most people recover after a few months.
Anti-GBM disease without lung involvement produces the same renal lesion as Goodpasture's disease. It is somewhat less common.
Masugi nephritis is an experimental anti-GBM disease. It is produced in rats by injections of anti-rabbit kidney anti-GBM antibodies prepared by immunizing rabbits with rat kidney tissue.
* Anti-GBM disease can also develop as a secondary phenomenon resulting to exposure to new GBM antigens in late MGN, late APSGN, and in Alport's syndrome patients (abnormal GBM) who receive kidney transplants.
In anti-GBM disease, immunofluorescence shows a diffuse linear pattern of antibody deposition along the GBM.
* Anti-GBM disease may have positive ANCA (usually p-ANCA) as well (reports of up to 1/3 of cases).

RPGN II: SEVERE IMMUNE COMPLEX DISEASE: Virtually any of these diseases can produce RPGN if it is severe enough.
"Post-infectious RPGN" is the severe form of post-streptococcal or other bacteria-based glomerulonephritis.
RPGN III: THE VASCULITIS SYNDROMES ("pauci-immune"):
{16832} segmental necrotizing RPGN, probably Wegener's
{16917} polyarteritis ("nodosa"???) in kidney, H&E
{16918} polyarteritis ("nodosa"???) , elastic stain
{16920} polyarteritis nodosa, angiogram
{16922} old polyarteritis ("nodosa"???), elastic stain
RPGN III (i.e., without significant immune deposits) is usually due to one of the systemic vasculitis syndromes, including Wegener's granulomatosis and variants, small-vessel polyarteritis, bad Churg-Strauss, bad rheumatoid arthritis with vasculitis (rare), and occasional cases of cryoglobulinemia (not so rare).
* Future kidney pathologists: Also remember lupus vasculitis, bad Henoch-Schonlein, and the vasculitis of subacute bacterial endocarditis. These are likely to produce immune deposits along with segmental necrosis (RPGN II).
All these vasculitis syndromes tend to produce a segmental necrotizing glomerulonephritis (SNGN) with crescents.
The majority will have anti-neutrophil cytoplasmic antibodies. If these are absent, there will still be neutrophils wrecking havoc: Am. J. Med. Sci. 340: 474, 2010.
* A kidney biopsy will probably not help you distinguish which vasculitis syndrome is present.
* Actual granulomas in or around the partially-necrotic glomerular tuft strongly suggest Wegener's or small-vessel polyarteritis, but are not pathognomonic. And if you see granulomas, the response to prednisone and cyclophosphamide will be good.
In patients in whom the diagnosis of Wegener's granulomatosis and/or polyarteritis (generalized or kidney-only) is known or suspected ("aggressive focal-segmental glomerulonephritis of unknown etiology, no immune deposits"), response to cyclophosphamide is usually excellent.
There is now a strong trend to treat all RPGN III / segmental necrotizing GN cases with cyclophosphamide and prednisone, and this practice has been supported recently by several pieces of evidence:
(1) It works (even in kids... J. Ped. 132: 325, 1998).
(2) Polyarteritis confined to the kidney is now a recognized entity. There seems to be no consensus as to whether to call this "nodosa", even if there are some little aneurysms.
(3) "Idiopathic RPGN III/SNGN" patients have the same anti-neutrophil cytoplasmic autoantibodies that characterize small-vessel polyarteritis (i.e., anti-myeloperoxidase) and Wegener's granulomatosis (i.e., anti-proteinase 3.)
Appallingly, RPGN is often missed by primary care physicians (at least in England) until it's too late. Check people's urine. See Br. Med. J. 301: 329, 1990.
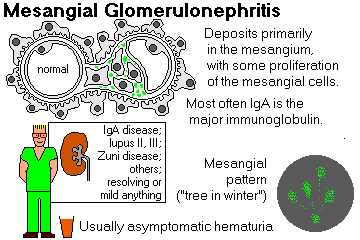
MESANGIAL PROLIFERATIVE GLOMERULONEPHRITIS ("mesangioproliferative GN", etc.)
This is an anatomist's diagnosis that covers a variety of relatively minor illnesses, including mild systemic lupus, resolving post-streptococcal glomerulonephritis, * rheumatoid arthritis, * maybe hepatitis B, etc.
Light microscopy shows only mesangial cell proliferation (i.e., more than five mesangial nuclei per tuft) and increased mesangial matrix. Immunofluorescence and electron microscopy show immune deposits in the mesangium in a majority of these cases.
For most of these patients, the prognosis is good.
Patients with IgA deposition have IgA nephropathy and have microhematuria and mild proteinuria.
Patients with C3-only have "C3-nephropathy" and have the same clinical picture.
* Patients with IgM deposition have "IgM nephropathy" and usually have more proteinuria and good response to treatment (try cyclophosphamide; or look for lupus Am. J. Med. Sci. 342: 530, 2011; common cause of steroid-resistant nephrotic syndrome in kids J. Clin. Path. 65: 1072, 2012). Patients with C1q-only have "C1q-nephropathy" and have the same clinical picture.
* There's an IgA/IgM/no C3 variant (Am. J. Clin. Path. 95: 863, 1991).
MEMBRANOUS GLOMERULOPATHY ("MGN", "membranous glomerulonephritis"; now it's"nephrotic idiopathic membranous nephropathy")
{16811} membranous glomerulopathy
{24849} membranous glomerulopathy, silver stain showing spikes
{00077} membranous glomerulopathy, H&E
{10565} membranous glomerulopathy
{16809} membranous glomerulopathy
{16807} membranous glomerulopathy, electron micrograph
{16808} membranous glomerulopathy, electron micrograph
{34237} membranous glomerulopathy, finely-granular immunofluorescence
{17015} membranous glomerulopathy, early
{17016} membranous glomerulopathy, early
{17017} membranous glomerulopathy, early, immunofluorescence
{17018} membranous glomerulopathy, early, electron micrograph
This reaction pattern is the commonest cause of nephrotic syndrome in adults (* most common in middle-aged men.)
Some patients have only mild proteinuria, and many of these recover completely. Around half of adults go on to chronic renal failure after 10-15 years of heavy, nonselective proteinuria.
Electron microscopy shows uniform, evenly-spaced subepithelial immune-complex deposits. Immunofluorescence shows a finely granular pattern of IgG (* almost all IgG4: Lancet 351: 670, 1998), C3, sometimes more.
The long-sought autoantigen in membranous GN seems to have been discovered. PLA2R is a normal compoment of the podocyte, and these patients have IgG4 antibodies against it (IgG4 aren't good compliment fixers, hence very little inflammation.) Exactly how the complexes form on the subepithelial surface is as mysterious as ever (NEJM 361: 11 & 81, 2009).
These deposits soon become incorporated into the GBM, making it look thicker on light microscopy (hence the name "membranous". Except in the earliest stage, spikes of GBM between the immune complexes are easy to see using PAS or silver stains.)
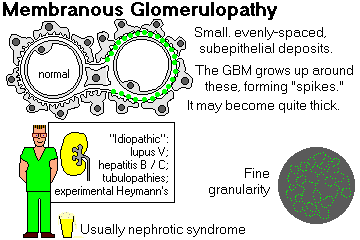
Most cases of membranous glomerulopathy are still "idiopathic."
Known causes include SLE, non-steroidal anti-inflammatory agents (this is
probably common
important: JAMA 276: 466,
1996), infections (hepatitis B virus -- this is probably the usual hepatitis B glomerulopathy;
update Arch. Dis. Child. 88: 446, 2003);
syphilis![]() ,
schistosomiasis
,
schistosomiasis![]() ,
Plasmodium malariae
,
Plasmodium malariae![]() , * sarcoid),
drugs (gold therapy and/or D-penicillamine for arthritis; captopril),
cow's milk (I was skeptical too but the bovine serum albumin's in the deposits:
NEJM 364: 2101, 2011),
de-novo after transplant, and
cancers (lung carcinoma and others.
, * sarcoid),
drugs (gold therapy and/or D-penicillamine for arthritis; captopril),
cow's milk (I was skeptical too but the bovine serum albumin's in the deposits:
NEJM 364: 2101, 2011),
de-novo after transplant, and
cancers (lung carcinoma and others.
* Contrary to what you may read elsewhere, "spike and dome" is an action-potential term, not a membranous glomerulonephritis term.
Still others result from renal tubules that have been damaged by some other disease, with deposition of RTE-anti-RTE antigen-antibody complexes in the deposits. RTE ("renal tubular epithelial antigen") comes from damaged tubules. You remember Heymann nephritis from your immunology course.
* We await confirmation of reports that many of these patients have antibodies against phospholipase A2 receptor (anti-PLA2R (NEJM 364: 689, 2011).
For treatment, various immunosuppressants have been used, with variable results. There's good short-term risk-benefit with rituximab (Lancet 360: 923, 2002). Tacrolimus plus glucocorticoids gets an 85% remission rate (best so far, very little toxicity): Am. J. Med. Sci. 339: 233, 2010. Lots of membranous glomerulonephritis stays stable or goes away by itself: NEJM 329: 85, 1993.
MINIMAL CHANGE GLOMERULOPATHY ("minimal change disease", "foot process disease", "lipoid nephrosis", "nil lesion", "nil disease")

{10566} foot process fusion, electron micrograph
{17032} foot process fusion, electron micrograph
{34294} foot process fusion, electron micrograph
The commonest cause of the nephrotic syndrome in children. Electron microscopy reveals diffuse loss of foot processes of epithelial cells. (Really the epithelial cells are swollen and this flattens the foot processes. This happens in many other glomerular diseases.)
There is no obvious evidence of immunologic disturbance, and the glomeruli appear normal by light microscopy.
The peak incidence is in children 2-3 years old, but adults may be affected.
Many of these adults will prove to have Hodgkin's disease, though most Hodgkin's patients get no glomerulopathy. The kidney problem will resolve when the malignancy is successfully treated.
Proteinuria is heavy but selective (i.e., mostly albumin is lost, unlike most glomerular diseases), and renal function remains good.
The long-term prognosis is excellent, often with dramatic response to corticosteroid therapy. For the idiopathic lesion in adults, it is not so good, and 5% of children have a chronic relapsing-remitting course that is likely to continue into adult life, though with relatively little morbidity or mortality (J. Ped. 147: 202, 2005).
The molecular pathology involves an unexplained loss of polyanions from the GBM, making it more permeable to albumin; nobody knows exactly how this causes foot process fusion, but it probably does.
* Puromycin and adriamycin ("the red death"; Kid. Int. 38: 851, 1990) each cause the disease in animals, and occasionally non-steroidal anti-inflammatory agents cause it in humans (see next lecture).
* In an animal model, minimal-change disease seems somehow to result from from the antiopoietin-like-4 protein on podocytes not being sialated. Glucocorticoid treatment stops the protein from beng expresed at all, and reverses the nephrotic syndrome (Nat. Med. 17: 117, 2011).
* An intractable case of recurrant minimal change disease in an adult clears with rituximab (Am. J. Kid. Dis. 49: 150, 2007). More on rituximab for difficult childhood nephrotic syndrome: Lancet 384: 1273, 2014.
FOCAL-SEGMENTAL GLOMERULOSCLEROSIS ("focal sclerosing nephropathy," "focal sclerosis")
{16802} focal-segmental glomerulosclerosis
{17035} focal-segmental glomerulosclerosis
{17036} focal-segmental glomerulosclerosis
{21028} focal-segmental glomerulosclerosis, with nice * hyalinosis lesion
|
|
Another pattern of glomerular injury that sometimes causes nephrotic syndrome in children and adults. It is considered a nonspecific response to non-immunologic or possibly-immunologic injury of the glomerular microvasculature.
The clinical picture and prognosis are less favorable than for minimal-change glomerulopathy (* with which it is often confused).
Patients typically have non-selective proteinuria, oliguria, hypertension, and progress to chronic renal failure despite steroid treatment after several years; much depends on the type. The disease is notorious for recurring in transplants. (* Hematuria is less common but may occur.)
Biopsy shows pretty-much-complete loss of all foot processes (as in minimal change disease) plus focal-segmental sclerosis and granular IgM and C3 in the sclerotic areas. Often there is actual detachment and loss of epithelial cells in some places; the sclerosis probably is where a group of podocytes fell off.
* There is usually striking hyaline arteriolar sclerosis.
* The splitters won, rightly, and the 2004 Columbia Classification now has five subtypes of FSGS with various meanings (Arch. Path. Lab. Med. 133: 217, 2009).
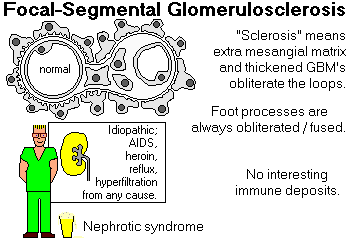
Most FSGS is "idiopathic", but there are several recognized causes.
* A few genetic loci involving podocyte proteins are known producing familial forms. Don't worry about them. For podocin / NPHS2, see J. Am. Soc. Nephro. 15: 832, 2004. CD2AP ("CD2-associated protein") -- Nephrol. Dial. Trnas. 24: 1858, 2009. For alpha-actinin-4, see Kid. Int. 70: 1054, 2006. TRPC6 is a calcium channel linked both to emotional illness and FSGS (Science 308: 1801, 2005). For Myo1E, see NEJM 365: 295, 2011. For INF2, an FSGS / Charcot-Marie-Tooth neuropathy locus, see NEJM 365: 2377, 2011. If the antioncogene WT1 is completely deleted on one chromosome, FSGS is likely whether or not there's also a Wilms tumor (Pediatrics 129: e1621, 2012).
AIDS nephropathy (Ann. Int. Med. 139: 214, 2003, great photos; also
Arch. Int. Med. 164: 333, 2004)
and most cases of heroin nephropathy
show FSGS, and
* pamidronate nephropathy and * schistosomiasis![]() and * lithium glomerulopathy often do, too. These tend to
produce an aggressive disease.
and * lithium glomerulopathy often do, too. These tend to
produce an aggressive disease.
Reflux nephropathy is considered a cause of FSGS, whether or not there is concurrent infection.
The "hyperfiltration lesion", seen whenever individual glomeruli must experience extra pressure because there's too little functionig kidney, begins with hyalinosis, and typically progresses to an FSGS-like lesion. First noticed in the mid-1980's, (Lancet 1: 1297, 1985, Nephron 43: 10, 1986; NEJM 316: 860, 1987), this discovery led to the phenomenally successful use of agents designed to slow the hyperfiltration lesion, forestalling the progression to end-stage disease of a majority of chronic kidney patients. It also explains today's preference for only partial nephrectomy ("nephron-sparing surgery") for small kidney cancers (Mayo Clin. Proc. 75: 1236, 2000).
* FSGS is very common in older rats (J. Geront. 49: B-157, 1994), including my own pets.
* In a large study of otherwise-unexplained FSGS, 2/3 of patients with primary FSGS, but not other glomerular disease, have elevated serum soluble urokinase receptor (suPAR); in a mouse model, it activates a podocyte integrin and effaces foot processes, causes proteinuia, and produses an FSGS-like histopathology (Nat. Med. 17: 952, 2011).
FSGS with staining of the podocytes for B7-1 (which is common) responds to abatacept, a biotech product that inhibits the protein (NEJM 369: 2416, 2013).
FSGS can be "secondary to obesity"; in this case or the loss of renal mass, it'll be the "not-otherwise-specified" type and be easy enough to keep at bay with ACE inhibitors. See below.
Regardless of cause, the prognosis is guarded. Glucocorticoids are of limited use for common FSGS, but work for AIDS nephropathy (at the price of additional immunosuppression, of course: Am. J. Med. 97: 145, 1994). The hyperfiltration lesion does quite well treated with ACE inhibitors with or without dietary protein restriction.
MEMBRANOPROLIFERATIVE "GLOMERULONEPHRITIS" (MPGN "type I", "mesangiocapillary glomerulonephritis type I", "cloverleaf disease", "membranoproliferative GN with immunoglobulin", etc.) Update MPGN I & MPGN II -- NEJM 366: 1119, 2012.
{08900} membranoproliferative glomerulonephritis, type I
{34264} membranoproliferative glomerulonephritis, type I
{17059} membranoproliferative glomerulonephritis, type I
{17060} membranoproliferative glomerulonephritis, type I
{16830} membranoproliferative glomerulonephritis, type I
{08902} membranoproliferative glomerulonephritis, type I
{16760} membranoproliferative glomerulonephritis, type I
{16761} membranoproliferative glomerulonephritis, type I
Another anatomist's diagnosis, not a specific disease. A pattern of immune-complex disease in which mesangial cells proliferate and send cell processes between basement membrane and endothelial cells. Watch for a subclassification soon (Thrombosis & Hemostasis 101: 271, 2009).
This gives the glomerular capillary walls a "double contour" or "tram track" appearance.

The mechanism of injury is immune complex deposition with activation of the classical complement pathway.
The immune-complex deposits are irregular in size and shape, located subendothelially, subepithelially, and mesangially, often extending from one region to another, contain IgG, C3, C4, C1q, etc.
Many cases exhibit "C3 nephritic factor" (an autoantibody that stabilizes alternate-pathway C3 convertase) in the serum. This causes excessive activation of complement, low C3 levels.
Although most cases are idiopathic, identifiable causes include infected shunts, SBE,
malaria![]() ,
schistosomiasis
,
schistosomiasis![]() ,
hepatitis B virus (again, Rx alpha-IF), hepatitis C virus (quite
common, big review NEJM 328: 465,
1993), sickle cell disease (Medicine 89: 18, 2010), heroin use, leukemia, lymphoma, SLE, bevacizumab treatment,
cryoglobulinemia (often with hepatitis C), C1-esterase inhibitor deficiency (Am. J. Kid. Dis. 19: 526, 1992).
,
hepatitis B virus (again, Rx alpha-IF), hepatitis C virus (quite
common, big review NEJM 328: 465,
1993), sickle cell disease (Medicine 89: 18, 2010), heroin use, leukemia, lymphoma, SLE, bevacizumab treatment,
cryoglobulinemia (often with hepatitis C), C1-esterase inhibitor deficiency (Am. J. Kid. Dis. 19: 526, 1992).
Idiopathic MPGN I most often affects young people, and presents variable clinical manifestations (nephrotic syndrome, nephritic syndrome, asymptomatic hematuria and proteinuria, sometimes RPGN.)
The long-term prognosis is guarded; various treatments have been tried without any spectacular successes.
{33331} good tram tracks
|
|
|
BASEMENT MEMBRANE DENSE DEPOSIT DISEASE ("membranoproliferative glomerulonephritis type II", "membranoproliferative GN without immunoglobulin", etc.)
{10568} dense deposit disease, immunofluorescence for C3
{16831} dense deposit disease
This pattern is most common in young people. It begins with asymptomatic hematuria or the nephrotic syndrome. Review Medicine 83: 18, 2004.
C3 nephritic factor is common as in MPGN type I; the whole business is still being sorted out. There isn't usually any immunogloblin deposited, but there is plenty of C3, usually as a"dense deposit".
Many of these patients have "partial lipodystrophy", with fat deposits disappearing from everywhere except the legs. Treating partial lipodystrophy with recombinant leptin (which seems to work) also helps the kidney problem (J. Clin. Endo. Metab. 89: 3199, 2004).
* The pig model features another deficient complement inhibitor: J. Clin. Invest. 95: 1054, 1995; molecules Am. J. Path. 161: 2027, 2002.
* This is one of the illnesses that has helped sort out the enormous complexity of the complement system -- for example, one component is made primarily by adipocytes. Review NEJM 344: 1058, 2001.
Light microscopy reveals proliferation of endothelium, thick GBM that includes a brown band on silver stain.
Dense band-like deposits within the GBM are visible on electron microscopy. (* The less-common "C3GN" has this stuff in the mesangium instead.)
Immunofluorescence shows these contain C3 but no C1q, C4 or Ig. Serum C3 is greatly reduced, while C4 remains normal. (The alternate complement pathway is being activated. C3NeF may be involved. Some patients have inborn deficiencies of, or mutations or, complement components.)
Historically, prognosis has been poor. There are now anecdotal accounts of complete remissions using eculizumab, the anti-C5 antibody (NEJM 366: 1161, 2012; confirmation NEJM 368: 2169, 2013).
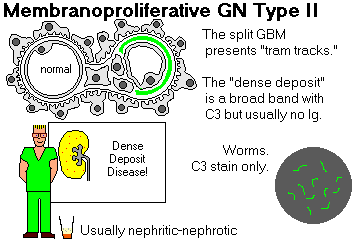
![]() Dense deposit disease
Dense deposit disease
Pittsburgh Pathology Cases
|
Pittsburgh Illustrated Case |
IgA NEPHROPATHY (see Medicine 73: 79, 1994).
{16751} IgA nephropathy, mesangial proliferation
{34297} IgA nephropathy, segmental proliferation
{16746} IgA nephropathy, mesangial immunofluorescence for IgA
|
|
Berger's (bare-ZHAY's) disease is now used to describe any idiopathic IgA nephropathy. (It formerly was applied to IgA nephropathy occurring in young men concurrently with a sore throat.)
Today "idiopathic IgA nephropathy" is the commonest serious glomerular disease. The pathogenesis continues to elude us. A single major gene is known (Nat. Genet. 26: 354, 2000). IgA1 (the deposited subtype) seems to be abnormally glycosylated (Lancet 369: 885, 2007), but beyond this, the disease is slow to yield up its secrets.
IgA nephropathy has a variable prognosis. There is still no effective treatment There are reports of good response to fish oil (NEJM 331: 1227, 1994). Glucocorticoids are reported to be of some benefit: Lancet 353: 883, 1999. Tonsillectomy for pediatric patients seems to be of benefit (Arch. Otol. 135: 85, 2009).
Patients have gross or microscopic hematuria, often with high blood pressure. The disease usually runs a chronic, slowly progressive course (Am. J. Med. 110: 434, 2001). Perhaps a third eventually develop renal failure.
Mesangial and focal-segmental proliferation and sclerosis may be seen by light microscopy. In bad cases, crescents develop.
Immunofluorescence shows IgA deposited in the mesangium (often with IgG, IgM, and/or C3, but no C1q or C4, i.e., the alternate pathway of complement is being activated.)
Serum IgA is often elevated, and IgA-containing immune complexes are often demonstrable, whether or not there is some primary disease to explain their presence.
*nbsp;The disease has about a 50% recurrence rate in renal allografts.
A glomerular lesion identical to that of Berger's disease occurs in some patients with cirrhosis / alcoholic liver disease, Zuni nephropathy?, celiac sprue (immune complexes from the gut bypassing the liver), in patients with other IgA-related problems (IgA-myeloma, dermatitis herpetiformis), in systemic lupus, and in hepatitis B carrier state (HBsAg-IgA complexes) It may resolve or go on to renal failure.
HENOCH-SCHONLEIN PURPURA (HSP, Lancet 369: 885, 2007): A fairly common syndrome most often occurring in children (more severe in adults) featuring:
All the signs and symptoms are attributed to immune-complex deposition in the vessels, joint spaces, etc.
The renal pathology is similar to that of idiopathic IgA nephropathy, and it shares the abnormal glycosylation of circulating IgA1.
* Both IgA nephropathy and Henoch-Schonlein IgA proteins are reported by one group to show specificity for streptococcal M-protiens. This awaits confirmation (Am. J. Path. 176: 608, 2010), but is consistent with the illnesses starting with a sore throat.
The BIG news in Henoch-Schonlein purpura is that a great many of these people carry the gene for familial Mediterranean fever (J. Ped. 143: 658, 2003). This correlates with many older reports of the two illnesses running together. This is now a robust finding, with various alleles here having different effects.
"CHRONIC GLOMERULONEPHRITIS"
{13374} "chronic glomerulonephritis"
A major cause of chronic renal failure in adults. An end-stage pool of glomerular diseases. (Remember, "chronic inflammation" is a time-honored misnomer for severe, irreversible scarring.)
About one third arise with no history of any of the well-recognized forms of early GN. ("Glomerular lesion too advanced to classify.")
Light microscopy shows hyaline obliteration of glomeruli, transforming them into acellular hyaline masses made of mesangial matrix, basement-membrane material, dense collagen, and trapped plasma protein.
Tubules are lost, vessel walls are thickened, and ultimately the kidney is totally destroyed.
Given the usual trans-stygian kidney, the pathologist cannot even tell whether the original disease was glomerular, interstitial, or vascular.
These patients suffer from uremia, get dialyzed, get transplanted, and eventually die.
SYSTEMIC LUPUS ERYTHEMATOSUS
{33195} wire loop
{16822} wire loop
{16824} wire loop, immunofluorescence
{16825} wire loop, fingerprints on EM
There is clinical evidence of glomerular involvement in a majority of SLE patients and renal failure is the cause of death in 30-40% of these people.
There is great variability in the morphology and clinical picture of lupus kidney disease. (Today, systemic lupus, not syphilis, is medicine's "great imitator".)
In all patients with suspected lupus, the kidney pathologist looks for tubular arrays ("reticular aggregates", "myxovirus-like inclusions" in endothelial cells), hematoxylin bodies (purple-staining nuclei of dead PMN's acted upon by anti-nuclear antibodies -- the term is passing out of use), "hyaline thrombi" (cryoglobulins, TTP thrombi, and/or red-wire loops that filled in), and organized "fingerprint" immune complex deposits.
"Hematoxylin bodies" have historically been considered specific for lupus. The others aren't. Further, knowing for certain what's a hematoxylin body on light microscopy is impossible, and if they are present, the diagnosis of lupus will be obvious clinically.
"Reticular aggregates" seen in the endothelial cells of lupus patients and HIV patients are not viruses, but alpha-interferon crystals.
The clinical manifestations depend on the histology, activity, and chronicity. These patterns change into each other as disease progresses, remits, and/or gets treated. Today, nephropathologists use the "Renal Pathology Society" classification (Kid. Int. 65: 521, 2004), which built on the World Health Organization's work.
Patients have mild proteinuria and sometimes microscopic hematuria.
You'll find immune-complex deposits around capillary loops in involved areas, with focal and segmental proliferation and sometimes necrosis.
* Hard-core pathologists subclassify Class III into III-A ("acute lesions" without scars), III-AC (acute lesions and scars), and III-C (scars without acute lesions.)
Patients have various degrees of proteinuria and hematuria. If this is the only lesion, patients may do well.
Massive mesangial, subepithelial, and/or subendothelial (red-wire loop) immune complex deposits with diffuse proliferation and sometimes even necrosis of glomeruli.
Patients typically have the acute nephritic syndrome. The prognosis is guarded, with around 30% ending up on dialysis within 15 years (worse than the others, Am. J. Kid. Dis. 19: 473, 1992).
* Again, hardcore pathologists have distinguished segmental (IV-S) and global (IV-G) involvement, with sub-subclassifications for the extent of scarring. Beyond noting whether the process is active or scar or both, the distinctions haven't proved very helpful in prognosticating or determining the best treatment: Neph. Dial. Tr. 23: 1298, 2007.
Immune complex deposition similar to other membranous GN.
Patients usually have the nephrotic syndrome. Often there is a mixed histologic picture. The prognosis is usually not good.
Be this as it may, most nephropathologists consider the "activity index" and "chronicity index" to be more useful as a guide to prognosis and treatment (for example, Arthr. Rheum. 33: 970, 1990).
The "activity index" (0-24) describes proliferation, necrosis, cellular crescents, hyaline thrombi, PMN's in the tufts, mononuclear cells in the interstitium.
The "chronicity index" (0-12) describes numbers of obsolescent glomeruli, fibrous (old burned-out) crescents, tubular atrophy, and interstitial fibrosis.
Of course, decreased complement levels (C3, C4) and increased levels of anti-double-stranded DNA are serologic evidence of active renal disease.
* Cyclophosphamide for lupus nephritis: Good, but no panacea (Lancet 340: 741, 1992). Fewer side effects and good results with mycophenolate mofetil: NEJM 343: 1156, 2000; update NEJM 353: 2219, 2005.

GLOMERULONEPHRITIS OF BACTERIAL ENDOCARDITIS
There is often an immune complex nephritis caused by deposition of circulating bacterial antigen-antibody complexes. (It is not caused by tiny emboli.)
The glomerular lesion ranges from focal GN to diffuse proliferative GN to MPGN I to crescentic GN.
Patients have varying degrees of hematuria, proteinuria, even RPGN.
DIABETIC GLOMERULOSCLEROSIS (huge review for pathologists: J. Clin. Path. 60: 18, 2007) Still probably the most common cause of end-stage renal disease.
{08892} KW disease
{17159} diabetes with hyalinized arteriole
{16789} diabetic glomerulosclerosis, electron micrograph (thick GBM)
{16790} diabetic glomerulosclerosis, electron micrograph (thick GBM)
{16791} diabetic glomerulosclerosis, H&E
{16792} diabetic glomerulosclerosis, H&E
{16793} diabetic glomerulosclerosis, H&E
{08893} Kimmelstiel-Wilson diabetic nodular glomerulosclerosis
{08895} Kimmelstiel-Wilson diabetic nodular glomerulosclerosis, PAS
{09877} Kimmelstiel-Wilson diabetic nodular glomerulosclerosis
{17158} Kimmelstiel-Wilson diabetic nodular glomerulosclerosis
{17171} end-stage diabetic glomerulosclerosis
|
|
|
|
|
All diabetics get hyperfiltration, thickened glomerular basement membranes and increased mesangial matrix (diffuse glomerulosclerosis; JAMA 263: 1954, 1990). In the old days, around 30% became detectable and these tended to progress to renal failure.
Both the hyperfiltration and the GBM thickening appear to be secondary to prolonged hyperglycemia.
The current principal suspect is advanced glycation (glycosylation) products turning on the gene to make basement membrane collagen (Proc. Nat. Acad. Sci. 91: 9519, 1994).
* However, the comparison with galactosemia -- where the GBM thickens but little else happens -- indicates that something else must be going on in diabetes.
Deterioration of glomerular function in diabetics can be slowed by controlling intrarenal hypertension with angiotensin converting enzyme inhibitors. Dietary protein restriction may also be useful. In any case, diabetic glomerulopathy is becoming much less of a new problem in insulin-dependent diabetics, thanks to tighter control (NEJM 330: 15, 1994). Today, among type I diabetics, only around 2% are in end-stage renal disease after 20 years, and only around 8% by 30 years (JAMA 2942: 1782, 2005). This is a triumph.
One's genetic makeup determines whether a diabetic will have slow or rapid deterioration of renal function. One key gene seems to be which allele you got for the angiotensin-converting enzyme gene itself (!! Diabetes 43: 690, 1994; J. Lab. Clin. Med. 131: 502, 1998; confirmation in knockout mice Proc. Nat. Acad. Sci. 98: 13330, 2001).
In many advanced cases, there is also nodular glomerulosclerosis ("Kimmelstiel-Wilson lesion"), with round masses of GBM-mesangial matrix material in the glomerular tufts.
This histologic picture is highly characteristic of diabetes, and it is usually present if the diabetes has been present for longer than 20 years (sometimes much less).
* There's only one mimic of nodular KW that might fool an experienced pathologist; it's IDIOPATHIC NODULAR GLOMERULOSCLEROSIS, (old work Am. J. Kid. Dis. 15: 281, 1990.) It occurs almost exlusively in people who smoke (J. Am. Soc. Nephro. 18: 2032, 2007) AND have longstanding hypertension, and is a ringer for nodular KW (even hyalinization of both afferent and efferent arterioles) EXCEPT that the endothelial antigens in the nodules are more abundant (i.e., new vessels formed and then got trapped: Neph. Dial. Trans. 21: 3571, 2006).
Many diabetics have albuminuria (occasionally in the nephrotic range), which progresses over years to renal failure (probably due to the mesangium choking off the glomerular capillaries).
* Future clinicians: Albuminuria over 30 mg/day predicts diabetic nephropathy, at least in type I diabetes. Check all diabetics annually. Albuminuria over 300 mg/day, hypertension, and falling GFR define the syndrome.
Diabetics also get severe atherosclerosis, arteriolar hyaline sclerosis, pyelonephritis,
staphylococcal![]() and candida
and candida![]() infections, papillary necrosis. All together, these are (not very usefully)
called "diabetic nephropathy." All about it: Kid. Int. 41(4), April 1992 (whole issue). Histopathology
correlating with decline in renal function: Diabetes 43: 1046, 1994.
infections, papillary necrosis. All together, these are (not very usefully)
called "diabetic nephropathy." All about it: Kid. Int. 41(4), April 1992 (whole issue). Histopathology
correlating with decline in renal function: Diabetes 43: 1046, 1994.
RENAL AMYLOIDOSIS (grading / scoring system Arch. Path. Lab. Med. 134: 532, 2010)
Amyloidosis A (SAA-derived) almost always involves the glomeruli, and amyloidosis AL (light-chain derived) often does, too. Amyloid is generally deposited first in the mesangium. Subepithelial (* "spike") and subendothelial deposits form later.
Telling amyloid AL from amyloid AA on H&E is impossible, and even the special stains are treacherous (Arch. Path. Lab. Med. 131: 917, 2007).
These patients often get the nephrotic syndrome. Often amyloidosis is discovered on kidney biopsy.
Eprodisate, the inhibitor of amyloid deposition, does seem to slow amyloidosis A of the kidney (NEJM 356: 2349, 2007).
* Leave the other "renal diseases with organized deposits" to us pathologists: Arch. Path. Lab. Med. 134: 512, 2010.
* "(Monoclonal) Light chain nephropathy" is a related, well-studied entity usually seen in patients with monoclonal gammopathies. Patients have proteinuria and/or renal failure. The light-chains are deposited just under the endothelial cells, causing cell slippage (?) and a light microscopic picture much like nodular diabetic glomerulopathy (except that the nodular are silver-negative and less PAS-positive). On staining, all basement membranes (GBM, Bowman's, tubular) will probably light up for the light chain.
* Non-amyloidotic fibrillary glomerulopathy is a cause of of nephritic-nephrotic syndrome; the fibrils are 20 nm diameter bits of polyclonal IgG.
* "Immunotactoid glomerulpathy" features 40 nm diameter tubules. By comparison, amyloid fibrils are 10 nm across. Updates Arch. Path. Lab. Med. 125: 534, 2001; Nephrol. Dial. Trans. 19: 2166, 2004.
* "Fibronectin glomeropathy" is a rare autosomal dominant illness ("FN1"), looks like silver-negative KW, and requires a special stain for diagnosis. Ditto the untreatable "heavy chain deposition disease": Am. J. Kid. Dis. 33: 954, 1999. Ditto "collagenofibrotic glomerulopathy": Am. J. Kid. Dis. 33: 123, 1999; Virch. Arch. 442: 163, 2003.
{10883} congo red stain of amyloid, polarized
{39804} amyloid in the glomeruli
THIN GBM DISEASE ("benign familial hematuria", "familial benign hematuria", "idiopathic essential hematuria, etc.")
A mild, very common family of illnesses, usually presenting as asymptomatic hematuria in childhood. The common known gene is a mild allele at an Alport's locus (COL4A5, one of the Type IV Collagen loci; J. Neph. 13: 15, 2000.) Female Alport's carriers are often affected (Hum. Path. 29: 404, 1998). There are also two other thin GBM loci that are dominant (COL4A3 and COL4A4; two doses gives an Alport's).
Renal biopsy shows patchy thinning of the GBM ("thin GBM disease"), without immune deposits or other abnormalities. Mild hearing problems are common. Pathology: Hum. Path. 33: 836, 2002 (as you'd expect there's a continuum from non-disease to Alport's with severe renal damage).
The group at Hopkins believes the prevalence of thin GBM disease in the asymptomatic population is about 1% (Arch. Path. Lab. Med. 130: 699, 2006); it may not involve all capillaries or all glomeruli (Arch. Path. Lab. Med. 130: 1533, 2006.)
CRYOGLOBULINEMIA... Worth knowing.
Cryoglobulinemia refers to the presence in the blood of marginally-soluble proteins (usually Ig's) that gel either in the cold or in the local hemoconcentration of a glomerulus. (Un-mixed cryoglobulins are generally M-proteins. Mixed ones tend to be antigen-antibody complexes of hepatitis B or C, or rheumatoid factor.) Hepatitis C "polyarteritis" with cryoglobulinemia is now infamous as a cause of renal disease (Am. J. Med. Sci. 331: 329, 2006).
The hallmark is the "pseudo-thrombus", a cryoglobulin plug in a capillary loop. Cryoglobulins also tend to produce subendothelial deposits ("red-wire loops") in glomeruli, and patients may get a glomerulonephritis (often membranoproliferative type I or diffuse proliferative) with proteinuria, hematuria, or even renal failure.
* Cryofibrinogens: Am. J. Clin. Path. 119: 114, 2003.
HEREDITARY GLOMERULONEPHRITIS (serious hereditary hematuria is generically called "Guthrie's disease")
The most familiar is the Alport's family, X-semi-dominant or autosomal-recessive nerve deafness plus progressive nephritis.
All the Alport's variants involve a defect in some collagen IV chain (Kid. Int. 43: 38, 1993; genetics Am. J. Path. 154: 1183, 1999; Am. J. Path. 160: 721, 2002; Lancet 375: 1287, 2010). Diagnosing this and thin-GBM disease: Arch. Path. Lab. Med. 133: 224, 2009. For some reason, the body shreds its own basement membranes (Am. J. Path. 169: 32, 2006); even more mysterious is the abundance of foam cells (lipid-laden macrophages) in the renal interstitim. Usually you'll do a skin biopsy first and ask the pathologist "Is this Alport's?" (Kid. Int. 55: 1217, 1999). Diagnosis by immunostaining: Arch. Path. Lab. Med. 125: 631, 2001. These easy molecular studies now render kidney biopsy unnecessary.
* There are a variety of other hereditary syndromes, including a hated version confined to Cyprus (founder effect) with a mutated complement protein (Lancet 376: 794, 2010).
| * Medical history buffs: Dr. Arthur Cecil Alport was a South African and a crusader for social justice. He resigned from the Royal College of Physicians in protest over the evils of colonialism and corruption, especially in Egypt. His book, "One Hour of Justice", was far ahead of its times. |
 Dr. Alport |
{24844} * Alport's syndrome, nice foam cells on the left
![]() Alport's
Alport's
Trichrome
WebPath Photo
* Medical students interested in the details about increased severity in men, focal-segmental sclerosis, basket-weave GBM with bizarre inclusions, mesangial foam cells, and so forth should consult standard texts. Guthrie's is a major cause of death in Samoyed dogs: Am. J. Path. 125: 536, 1986. Doggy Guthrie's: Am. J. Path. 139: 751, 1991
* What's a much more important cause of renal failure plus nerve deafness? Aminoglycoside toxicity!
* Nail-patella syndrome is autosomal dominant with variable penetrance; it features tiny kneecaps, hypoplasia of some or all of the nails, and a distinctive glomerular lesion (chunks of type III collagen) that usually is mild. The mutant gene is one that helps define structures, LMX1B (Am. J. Hum. Genet. 63: 1651, 1998).
* Mutant beta-2 laminin causes Pierson syndrome, a congenital nephrotic syndrome (Pediatrics 118: e501, 2006). Entactin / nidogen-1 is another glycoprotein presently in search of a genetic syndrome.
Remember:
The most common cause of protein in the urine is orthostatic proteinuria, from renal venules that ooze protein when hydrostatic pressure is high.
Trace proteinuria almost never deserves a special workup. It is the rule in people with chronic microvascular disease (i.e., high blood pressure, diabetes without glomerular disease, old age), and can be taken as a reasonable measure of "poor protoplasm".
The major causes of nephrotic syndrome:
The most common cause of urine suddenly turning red is exercise, especially pavement pounding ("march hemoglobinuria," "jogger's hemolysis", also exertional myoglobinuria).
Ask about beet and/or blackberry ingestion too; some people have a hereditary inability to metabolize some pigment.
Focal-segmental glomerular lesions are seen in focal segmental glomerulosclerosis, mesangial proliferative glomerulonephritis (a few cases, focal sclerosis), IgA nephropathy (Berger's, Henoch-Schonlein, a few SLE's), vasculitis (Wegener's, small-vessel polyarteritis, SBE, some SLE's), * Alport's, * and a few Goodpasture's.
Fabry's is the only common storage disease with significant renal involvement. You'll learn about the electron microscopic "zebra bodies" in genetics.
We'll learn about the lesions of ECLAMPSIA when we study women's problems.
KIDNEY MALFORMATIONS
Birth defects:
Agenesis
![]() Agenesis of the kidneys
Agenesis of the kidneys
WebPath Photo
Hypoplasia: a kidney with fewer than five lobules and calyces
Pelvic kidney (kinked ureter can result in poor drainage, stones, and infection); some of these are midline "cake kidneys" that never separated.
Horseshoe kidney: present in 1/500 or so autopsies, fusion of upper or lower poles of the kidneys. Seldom a problem as long as the ureters are not compressed badly to produce infection and/or stones.
{16972} horseshoe kidney
|
|
![]() Nephroblastomatosis
Nephroblastomatosis
Rare malformation
Wikimedia Commons
RENAL TUBULAR DYSGENESIS is a rare, lethal syndrome with kidneys that look normal grossly but have extreme hypoplasia of the proximal tubules.
Non-genetic causes of the same kidney picture are ACE-inhibitor use by the mother, and twin-twin transfusion syndrome (Path. Res. Pract. 196: 861, 2000).
|
|
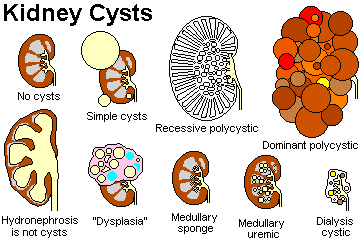
Abnormal differentiation / hyperplasia of tubular elements leads to the cystic diseases of the kidney (Kid. Int. 33: 8, 1988 -- * by my late teacher, Dr. Frank Carone, a leader in this field.) Today, there are at least 33 genes known in the cystic disease family, with many more awaiting discovery (Arch. Path. Lab. Med. 134: 569, 2010).
Apart from "cystic renal dysplasia", these diseases feature tubules whose cells proliferate to form cysts, which are involved with remodelling of the interstitium.
CYSTIC RENAL "DYSPLASIA" ("Potter II"; today, "multicystic dysplastic kidney", or "obstructive renal dysplasia" if hydronephrosis is present): persistence of primitive mesenchyme, which may produce cartilage, undifferentiated mesenchyme, and immature collecting ductules
This is the most common cause of an abdominal mass in newborns. Most are "idiopathic"; a few genes are known that produce a multicystic dysplastic phenotype without obvious obstruction (Hum. Mol. Genet. 15: 2363, 2006; Am. J. Kid. Dis. 47: 1004, 2006).
Regardless of phenotype, most cases probably result from failure of glomeruli to drain into the ureter during embryonic life (Virch. Arch. 439: 560, 2001; Ped. Int. 45: 605, 2003). There may be subtle failure of the ureteric bud to meet the blastema, or the lower urinary system may be malformed. Review Am. J. Kid. Dis. 32: 535, 1998. Update Kid. Int. 69: 190, 2006.
{10563} renal multicystic dysplasia (this is NOT polycystic kidney)
{10241} renal multicystic dysplasia
{39684} renal multicystic dysplasia; find the cartilage
{08453} renal multicystic dysplasia
{21006} renal multicystic dysplasia (this is NOT polycystic kidney or tumor)
![]() Multicystic dysplastic kidney
Multicystic dysplastic kidney
WebPath Photo
AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE ("adult polycystic kidney disease", "ADPKD", * "Potter III" in the old classification): NEJM 329: 332, 1993; Am. J. Kid. Dis. 28: 788, 1996; nice picture NEJM 333: 31, 1995).
Common (500,000 cases in the U.S.) autosomal dominant disease with complete penetrance (1 in 800 people or so).
Usually, the disease is on chromosome 16p1.3 (PCKD 1 locus) where it codes for "polycystin 1", a tubular organizer (Proc. Nat. Acad. Sci. 93: 1524, 1996); polycystin 2 has been identified as an integral membrane protein (PCKD2 locus, Science 272: 629, 1996, milder but not benign Lancet 353: 103, 1999). Prognosticating adult polycystic kidney disease: NEJM 323: 1085, 1990 (still good; if your ultrasound is normal as a young adult, you probably don't have it).
Hundreds of cysts, measuring up to 4 cm in diameter, develop from all levels of nephron including Bowman's capsule. As they form, the surrounding normal kidney cells undergo apoptosis (NEJM 333: 18 & 56, 1995).
By the time the patient is forty years old, the kidneys are often the size of footballs. Surprisingly, they may still be working. Half of these patients are on dialysis or transplanted by age 70 or so.
Patients typically get high blood pressure as adults, years before renal failure develops. Many die of ruptured berry aneurysms (management of the prospective berry aneurysm patient with PCKD: NEJM 327: 916, 1992).
Cyst infections by bacteria are hard to spot and hard to treat.
About a third of these patients have hepatic cysts too, which can be a nuisance if they make the liver too large. A few have cysts in the pancreas. You'll spot these on CT scan.
* A good deal is written about the molecular pathways of cyst formation and so forth. A vasopressin receptor antagonist prevents the formation of cysts (Nat. Med. 10: 363, 2004). The everolimus trial slows that the drug slows the enlargement of the kidneys without slowing their functional impairment (NEJM 363: 830, 2010). The sirolimus study showed no benefit at all (NEJM 363: 820, 2010).
{00056} adult, autosomal-dominant polycystic kidney (26 lb!)
{00059} adult, autosomal-dominant polycystic kidney (same case as 56)
{10553} adult,
autosomal-dominant polycystic kidney
{05965} adult, autosomal-dominant polycystic kidney, histology
|
Australian Pathology Museum Hey, how about a ruler?
|
![]() Adult Polycystic Kidney Disease
Adult Polycystic Kidney Disease
CDC
Wikimedia Commons
AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE ("childhood" or "infantile" polycystic kidney disease", "ARPKD"; "Potter I"; update Pediatrics 111: 1072, 2003; spectrum and forme frustes Medicine 85: 1, 2006)
Rare autosomal recessive disease (PKHD1/fibrocystin) with huge, white, smooth-surfaced kidneys. Cysts 1-2 mm in diameter develop from the collecting ducts; they are arranged in a radial, "sun-ray" pattern perpendicular to the capsule (because the collecting ducts are dilated). These kidneys are huge at birth.
This is normally fatal in infancy or early childhood; typically, the enormous kidneys restrict the ability of the lungs and gut to function. Many children also have congenital portal fibrosis of the liver (Gastroent. 144: 112, 2013).
* Keeping these babies alive may require cutting out one kidney to preserve room for the rest of the abdominal organs (there's a bit of renal function remaining, at least at birth): J. Ped. 127: 311, 1995.
* PKHD1 is a cilia protein. Nobody understands how it leads to polycystic kidneys.
{16978} infantile, autosomal-recessive polycystic kidney disease
{17223} infantile, autosomal-recessive polycystic kidney disease
{15817} infantile, autosomal-recessive polycystic kidney disease (huge
kidneys)
{15818} infantile, autosomal-recessive polycystic kidney disease
{17224} infantile, autosomal-recessive polycystic kidney disease, histology
|
|
![]() Autosomal recessive PCKD
Autosomal recessive PCKD
WebPath Photo
MEDULLARY SPONGE KIDNEY
Very common (1 in 200 people) idiopathic process. Dilated distal portions of collecting ducts superficially resemble cysts.
Usually it is just a radiologist's curiosity. Sometimes stones form in the "cysts", and this entity is in the "differential" of chronic back pain.
{25322} medullary sponge kidney, sketch
UREMIC MEDULLARY CYSTIC DISEASE / "nephronophthisis"
A group of diseases, seemingly genetic, with cysts at the corticomedullary junction and severe damage to the cortex.
* The hereditary forms include three diseases coded by NPHP1 ("nephrocystin"), NPHP2, and NPHP3, which are the most common causes of endstage renal disease in children and young adults (genes Proc. Nat. Acad. Sci. 98: 9836, 2001; update Nat. Genet. 34: 455, 2003.) Another severe autosomal recessive is at XPNPEP3, which codes a mitochondrial protein important in "primary cilia" (they are ubiquitous and don't beat; J. Clin. Inv. 120: 660 & 791, 2010).
* Milder, autosomal dominant forms are carried at the MCKD1 and MCKD2 loci (Kid. Int. 64: 788, 2003; Kid. Int. 68: 1472, 2005). XPNPEP3: J. Clin. Inv. 120: 791, 2010.
As with most diseases that begin in the medulla, the initial manifestations are inability to retain sodium and water.
{16985} medullary cystic disease, the bad kind, gross;
it has been in formalin for a long time and lost its color
{25323} medullary cystic disease, the bad kind, sketch
{25324} medullary cystic disease, the bad kind, gross
|
* Glomerulocystic disease, with dilated Bowman capsules, is secondary to some other illness, famously tuberous sclerosis; you will not be asked to recognize it.
* OTHER CYSTIC DISEASES
"ACQUIRED DIALYSIS CYSTIC DISEASE" ("trans-stygian kidney")
This is a misleading name for the kidneys in people who have been kept alive for a long time on dialysis.
The kidneys are useless nubbins with a few remaining tubules stretched wide open ("cysts").
In addition to scar tissue and a few chronic inflammatory cells, the pathologist may find squamous metaplasia of glomerular epithelium, oxalate crystals in the tubules, fibromuscular masses in the blood vessels, and cortical adenomas and renal cell carcinomas (J. Urol. 151: 129, 1994; radiologists enjoy Radiology 195: 667, 1995).
These kidneys can develop stones, painful bleeding, and/or aggressive carcinomas (not rare; Am. J. Kid. Dis. 16: 452, 1990).
We do not really know why this happens to the kidney, but it also occurs in patients who have not been dialyzed. Most plausible seems to be the idea that the underlying problem is the extreme vascular narrowing, with cells dying off when perfusion drops slightly and then proliferating again when minimally-adequate circulation is restored. The vessels in badly-damaged kidneys become extremely narrowed, and trans-stygian kidneys can develop behind a single stenotic renal artery.
* "Trans-stygian" refers to the river Styx that the dead crossed in Greek mythology.
{38869} acquired dialysis cystic disease" (trans-stygian kidney)
|
|
|
SIMPLE CYSTS
![]() Simple renal cysts
Simple renal cysts
WebPath Photo
A few cysts in a kidney (especially in an old person) is one of the commonest incidental findings at autopsy. These often develop after small kidney infarcts ("arterial nephrosclerosis"). No danger to health.
These keep turning up on scans and causing consternation, but the distinction from the more serious cystic diseases is obvious (Am. J. Roent. 176: 843, 2001).
* Tuberous sclerosis patients often have several simple cysts; this is perhaps the least of their many problems. Animal model Am. J. Path. 162: 457, 2003.
* Leave the diagnosis of "multilocular cyst" ("cystic nephroma", a curious hamartoma) to us. It looks like localized renal dysplasia but supposedly has no solid areas, or a benign, multicystic, sharply-demarcated tumor made of Wilms-type cells. Children or adults.
DISORDERS OF THE PROXIMAL TUBULE ("Fanconi" and so forth)
CONGENITAL: INBORN ERRORS OF TRANSPORT
Renal glycosuria (despite normal serum levels)
* Hypophosphatemic vitamin-D resistant renal rickets: X-linked dominant phosphate wasting syndrome.
Renal transport aminoacidurias (blood levels normal, but proximal tubules cannot reabsorb one or more amino acids from the glomerular filtrate)
* Renal glycinuria: glycine, proline, and hydroxyproline are lost
* Hartnup disease: most neutral amino acids are lost
Cystinuria: cystine, * lysine, * arginine, and * ornithine are lost. (Common. These patients get cystine kidney stones, and kidney damage from cystine crystals.)
Congenital Fanconi syndrome (generalized proximal tubular dysfunction): bicarbonate, phosphate, glucose, amino acids, calcium, potassium all lost (* other problems too). The major concern is bone disease. (* "Fanconi's anemia", a different problem, involves genetically programmed marrow failure.)
ACQUIRED FANCONI SYNDROME
Poisoning: lead (Am. J. Med. Sci. 327: 341, 2004; it's still around), cadmium, bismuth, oxalate, outdated tetracycline, * phenol, * glue sniffers
* A European team claims that subclinical Fanconi's due to subtle cadmium poisoning is commonplace: Lancet 336: 699, 1990. During the 1990's, there was a pollution scare in Europe (Lancet 343: 1523, 1994). On follow-up, these folks are having no problems with kidney dysfunction (Lancet 354: 1508, 1999), and even the discoverers aren't worried.
* Likewise, many children growing up in the horribly metal-polluted Aral Sea region do have measurable impairment of tubular function, some areas worse than others (Arch. Dis. Child. 88: 966, 2003). But the responsible pollutants could not be identified, and there's nothing to suggest the affected children are less healthy. Stay tuned.
Systemic disease: Wilson's disease (copper overload in the proximal tubule causes it to malfunction), plasma cell myeloma, * galactosemia (the bad variant), * cystinosis (a lysosomal storage disease, not to be confused with simple cystinuria).
| * Medical history buffs: Dr. Fanconi, one of the great pediatricians of the 20th century, was a bio-politician whose primary concern in later years was the health of children in the poor nations of the world. |
 Dr. Fanconi |
* GENETIC DISEASE OF THE TUBULES: Nat. Genet. 13: 183, 1996; Am. J. Med. Sci. 322: 316, 2001
Bartter's... Defective Na-K-2Cl cotransporter in the ascending limb; low potassium, metabolic alkalosis, normal blood pressure (Nat. Genet. 12: 24, 1996). This'll give you hyperplasia of the JGA -- why?
Gitelman's... Super-avid Na-Cl cotransporter in the distal tubule; low calcium, low potassium, metabolic alkalosis
Liddle's... Super-avid Na-retainer/K-waster in the distal portions of the nephron; bad hypertension: NEJM 330: 178, 1995; Am. J. Med. Sci. 322: 302, 2001.
We have already mentioned the renal tubular acidosis syndromes, and of course some are genetic.
Carbonic anhydrase II deficiency gives mental retardation and distal renal tubular acidosis (J. Ped. 132: 717, 1998).
ACUTE TUBULAR NECROSIS ("ATN", the usual cause of what clinicians call "acute renal failure", "renal shutdown", nowadays "acute renal injury", "acute kidney injury", etc.) NEJM 334: 1448, 1996; updates Chest 128: 2847, 2005; JAMA 294: 813, 2005, J. Clin. Inv. 121: 4210, 2011.
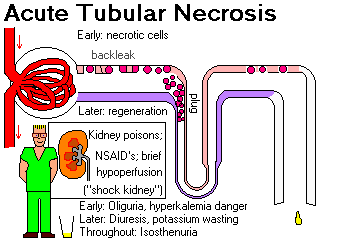
{07026} mercury coagulation necrosis![]() of the proximal tubule (the
dead cells have undergone dystrophic calcification)
of the proximal tubule (the
dead cells have undergone dystrophic calcification)
{32072} mercury necrosis of the proximal tubule
{17106} acute tubular necrosis, mitotic figure
{10262} acute tubular necrosis
|
|
|
|
|
|
|
Acute tubular necrosis is the designation for all forms of acute renal failure caused by damage to tubular epithelial cells.
ISCHEMIC ATN: caused by ischemia (shock, i.e., massive hemorrhage, septic shock, neonatal asphyxia -- J. Ped. 158-2S: e29, 2011), etc.; remember you can be in shock without being hypotensive!) Also seen in new transplants, from contrast media, even the "safe" ones (Br. J. Rad. 71: 357, 1998; no one understands the pathophysiology of "contrast nephropathy" NEJM 354: 379, 2006; hard to prevent / treat Br. J. Rad. 86: 20120272, Jan 2013; this is one of the banes of emergency percutaneous coronary interventions in folks having heart attacks -- Am. J. Card. 105: 624, 2010; even worse if the patient also just had heart surgery: Ann. Thorac. Surg. 95: 513, 2013); in low-output states, non-steroidal anti-inflammatory agents and ACE-inhibitor drugs, etc.
NEPHROTOXIC ATN: caused by a wide variety of renal poisons, including heavy metals (notoriously mercury), organic solvents (carbon tetrachloride, ethylene glycol), or antimicrobial agents (best-known are gentamicin and amphotericin B).
{07201} he died of ethylene glycol drinking
* Also: Renal failure among partakers of a meal consisting largely of raw carp gallstones ("pride in our ethnic cuisine"): JAMA 274: 604, 1995. Followed by acute hepatorenal failure in four partakers of a meal consisting largely of snake gallbladders (Clin Tox. 44: 387, 2006).
A vitamin C abuser ruins a transplanted kidney: NEJM 358: e4, 2008.
* Also: The Bangladeshi pharmaceutical manufacturer that put ethylene glycol in their Tylenol elixir and killed a lot of people. (Do you think it's "Uncle Sam" or "those tort layers" who keep this kind of thing from happening in the U.S.? I doubt that it's "big business's smarts and virtue"....) Br. Med. J. 311: 88, 1995.
* A cult drinks huge amounts of a copper salt solution and they end up with massive hemolysis and renal failure: Am. J. Med. 98: 311, 1995.
* "Chinese holistic natural herbal arthritis remedy" (adulterated with huge amounts of mefenamic acid, no surprise) causes renal failure: Arch. Int. Med. 155: 211, 1995.
PIGMENT NEPHROPATHY: massive hemolysis (red cell stroma plugs the tubules; ABO mismatch, falciparum
malaria![]() , snakebite, others),
massive rhabdomyolysis (current review NEJM 361: 62, 2009;
many articles that are good reads... overexertion Ann. Emerg. Med. 23: 1301, 1994,
crush injury, lightning, cocaine (Arch. Path. Lab. Med. 131: 1817, 2007),
beatings South. Afr. Med. J. 37: 214,
1994; torture Nephron 63: 434, 1993 -- myoglobin and other muscle debris plugs the tubules etc. --
Arch. Int. Med. 148: 1553, 1988; Br. Med. J. 298: 443, 1989, J. Traum. 38: 252, 1995, a famous "does
THAT count as torture?" case in the Middle East), etc.
, snakebite, others),
massive rhabdomyolysis (current review NEJM 361: 62, 2009;
many articles that are good reads... overexertion Ann. Emerg. Med. 23: 1301, 1994,
crush injury, lightning, cocaine (Arch. Path. Lab. Med. 131: 1817, 2007),
beatings South. Afr. Med. J. 37: 214,
1994; torture Nephron 63: 434, 1993 -- myoglobin and other muscle debris plugs the tubules etc. --
Arch. Int. Med. 148: 1553, 1988; Br. Med. J. 298: 443, 1989, J. Traum. 38: 252, 1995, a famous "does
THAT count as torture?" case in the Middle East), etc.
VIRAL HEMORRHAGIC FEVERS often cause temporary renal shutdown: J. Inf. Dis. 181: 1964, 2000. In Europe, voles carry Puumala hantavirus that produces "nephropathia endemica" (Scand. J. Inf. Dis. 32: 125, 2000; now "hemorrhagic fever with renal syndrome / HFRS" (J. Clin. Endo. Metab. 93: 2722, 2008); complete recovery is the rule (Kid. Int. 69: 2043, 2006).
The pathophysiology of ATN is complex:
In ATN, the renal output is normal or low; the urine is iso-osmotic with sodium concentration close to that in the glomerular filtrate.
* Future ward physicians: ATN due to aminoglycoside antibiotics -- gentamicin, etc. -- is usually non-oliguric. It is common.
The histology reflects the pathophysiology.
Ischemic ATN: you may see a few necrotic cells or denuded basement membrane, but they are rare. Look for dilated tubules and interstitial edema (from backleak). Proximal tubular cells may appear flattened (* "simplified" from loss of the apical cytoplasm). Proximal tubular cells show evidence of regeneration at all stages (basophilic cytoplasm, active nuclei, maybe even a mitosis or two).
* Electron micrographs show disruption of the basolateral infoldings and brush borders.
* Future pathologists: Unlike post-mortem decomposition / autolysis, when there has been ATN in life, the cells still form coils and ribbons. Old observation recently proved: J. For. Sci. 54: 439, 2009.
Nephrotoxic ATN: frank necrosis is seen, usually limited to the proximal tubules, without rupture of basement membranes.
Clinical picture of ischemic acute tubular necrosis:
Onset during a medical or surgical catastrophe
Oliguric stage, with urine production 50-400 mL/24 hr, and isotonic (hyperkalemia is the major danger).
Diuretic stage, with rapid loss of fluid and potassium (infection and hypokalemia are the major dangers). The tubules have regenerated during this phase (so there is no backleak), but they are not yet doing their job of reabsorbing the glomerular filtrate.
Unless your patient is in deep coma, has cancer, has persistent severe hypotension, and/or needs mechanical ventilation, he or she will probably recover. Today, "people die with ATN, not of ATN".
The same findings as in the above (upbeat) article were duplicated in a large, impressive study with a much more doleful tone and a focus on recognizing bad prognosis, i.e., "managed care and rationing have come to the U.S." Enjoy Arch. Int. Med. 155: 1505, 1995. In grave illness, kidney failure is a herald of death, though not today the actual cause.
Other causes of acute renal shutdown, other than ATN, include:
Also, rule out prerenal causes, i.e., dehydration, shock / hemorrhage and congestive heart failure, and postrenal causes, i.e., urinary tract obstruction by tumors, blood clots, prostate trouble. Review of renal failure in CHF: Postgrad. Med. 95(8): 141 & 153, Jun. 1994. And if course, "if the patient isn't putting out any urine", be sure the Foley isn't just kinked / plugged / blocked.
HEPATORENAL SYNDROME (Med. Clin. N.A. 93: 855, 2009; Dig. Dis. Sci. 57: 210, 2012 reaffirms its untreatability in the absence of liver transplantation as does Gut 62: 131, 2013).
Kidney failure that develops in patients with liver failure, without anatomic changes in the kidney (except perhaps bile-staining of hyaline casts). Often this follows enthusiastic administration of high-powered diuretics ("Lasix," etc.) to a cirrhotic "to help with the ascites".
The pathophysiology is getting worked out.
Liver failure from any cause produces hypotension, and "hepatorenal syndrome" may also have a component of "shock kidney" (acute tubular necrosis).
But unlike classic ATN, urine sodium is low (less then 10 mEq/L), while urine osmolality tends to be around 100 mOsm higher than serum. And old claims that hepatorenal syndrome simply reflects diminished "effective circulating plasma volume" ignored the observation that volume expansion doesn't help these patients.
The evidence nowadays points to dilation of the small arteries in the splanchnic bed and inappropriate constriction of the small arteries in the kidney, due to things in the blood that aren't being properly cleared by the liver. Today's usual suspects are endothelin 1 (Gut 53: 159, 2004) and some of the prostaglandins.
A while back, vasopressin analogues were tried by optimists for this famously-intractable problem. Reviews Gut 47: 288, 2000; Postgrad. Med. 109: 91, 2001; Gastroenterology 122: 923, 2002.
PYELONEPHRITIS AND OTHER UPPER URINARY TRACT INFECTIONS
An extremely common, serious problem in clinical medicine.
Causes:
Ascending infection (E. coli and other gram-negatives, swimming upstream from bladder infections, common)
Hematogenous infection (especially staph![]() and TB
and TB![]() , uncommon)
, uncommon)
{39794} acute pyelonephritis, dilated and inflamed pelvis
{17177} acute pyelonephritis, histology;
note the neutrophils, sparing the glomeruli
{10564} acute pyelonephritis, gross;
little abscesses
{16868} acute pyelonephritis, gross
{08876} acute pyelonephritis, histology
{08877} acute pyelonephritis, histology
{16866} acute pyelonephritis, histology
{17574} acute pyelonephritis, gross
{17575} acute pyelonephritis and hydronephrosis
{25332} acute pyelonephritis with hydronephrosis
{25335} acute pyelonephritis, histology; tubule
full of polys
{00071} renal abscess and kidney stones
|
|
|
|
ACUTE PYELONEPHRITIS
Predisposing conditions:
(up to 40, females predominant; over 40, males predominant because of big prostates)
As expected, PMN's infiltrate the interstitium and tubules. If you see even 1-2 polys in tubules, you can probably call it acute pyelonephritis.
Patients have fever, pain at costovertebral angle, PMN's and white cell casts in the urine.

Papillary necrosis is a dreaded complication of acute pyelonephritis that occurs mostly in diabetics.
* The papillae die all at once. (Contrast analgesic nephropathy, in which papillae die one at a time.)
* Beethoven apparently had papillary necrosis, perhaps from his high intake of a salicylate powder for his hangovers: J. Roy. Soc. Med. 86: 159, 1993; Am. J. Kid. Dis. 21: 643, 1993.
{16950} papillary necrosis in phenacetin abuse;
necrotic patches are greenish
{16894} papillary necrosis, same picture backwards! You need a laugh right about now.
{21015} papillary necrosis in pyelonephritis
CHRONIC PYELONEPHRITIS (includes reflux nephropathy)
Because of changing concepts and criteria for the diagnosis of chronic pyelonephritis, the frequency markedly decreased.
By time-honored convention, "chronic pyelonephritis" refers to any chronic renal infection. (Non-infectious inflammation of the renal interstitium is called "interstitial nephritis").
By time-honored misnomer, "chronic pyelonephritis" also is used to describe severe scarring from one or more healed kidney infections. Pyelonephritis always produces some renal scarring around the calyces and pelvis and among the tubules.
At autopsy, the pathologist looks for broad, U-shaped scars over distorted calyces. The U-shaped scars result from healed abscesses, which are superficial and wide -- they can look like old acne scars. The old infection is never evenly-distributed. The scarring is most likely to be worst at the renal poles (pressure physics), but can be anywhere.
Microscopically, this a patchy process with periglomerular fibrosis (outside Bowman's capsule) and interstitial scarring. (Scar retraction produces dilatation of cast-filled tubules, or "thyroidization." By this time, of course, the white cells are long gone, so don't expect to see white cells in the protein casts.)
The clinical picture varies:
Note that chronic pyelonephritis is an extremely important cause of morbidity and mortality in the wheelchair-bound.
{16883} "chronic pyelonephritis", gross
{08868} "chronic pyelonephritis", trichrome, blue is scar
{08881} "chronic pyelonephritis", lymphocytes
{08883} "chronic pyelonephritis", lymphocytes
{08884} chronic pyelonephritis", lymphocytes. I bet this is really chronic autoimmune interstitial
nephritis because of all the lymphocytes.
{17185} "chronic pyelonephritis", thyroidization
{16871} "chronic pyelonephritis", old scars, gross
{08731} chronic pyelonephritis, old scarring
{16872} "chronic pyelonephritis", old scars
{21011} "chronic pyelonephritis", gross
{28934} "chronic pyelonephritis", gross
{28937} "chronic pyelonephritis", good example, glomeruli spared, tubules shot
{28940} "chronic pyelonephritis", severe tubular atrophy
{28946} "chronic pyelonephritis", periglomerular fibrosis
|
|
XANTHOGRANULOMATOUS PYELONEPHRITIS (Arch. Path. Lab. Med. 135: 671, 2011) is a special, common type of chronic pyelonephritis, usually in folks who have had several episodes of acute pyelonephritis.
Located mostly around the pelvis, the process tends to spread, and it can grow out around the kidney and may erode into the area around the kidney and even produce fistulas to the groin, back, or gut.
Lipid-laden foamy-looking macrophages form large yellow nodules that look very much like renal cell carcinoma, both grossly and microscopically.
{24044} xanthogranulomatous pyelonephritis, gross, trust me
{11541} xanthogranulomatous pyelonephritis, good foam cells
{24045} xanthogranulomatous pyelonephritis, good inflammation and foam cells
Other causes of ACUTE INTERSTITIAL NEPHRITIS (presumably immune, in the absence of bacterial infection) include lupus, drug reactions (a few are notorious; they start 1-2 weeks after the beginning of therapy) and other "autoimmune" problems that generate anti-TBM antibody. These are just now being worked out, but are an important cause of renal failure. On biopsy, be alert for "tubulitis" (lymphocytes in the tubular epithelium).
{16892} anti-tubular basement membrane auto-antibody fluorescence
* Future ophthalmologists: There is a curious link between acute interstitial nephritis, due to systemic disease or "idiopathic", and inflammation of the iris (uveitis).
The BK POLYOMAVIRUS is ubiquitous (* 90%
of adults are seropositive) and causes an interstitial nephritis in the immunosuppressed: NEJM 326: 988, 1992 --
especially kidney transplant patients. Pathologists look for "decoy cells" with
huge basophilic intranuclear inclusions![]() and "comet cells" (short cytoplasmic tails);
we now have PCR too (Arch. Path. Lab. Med. 123: 807, 1999;
NEJM 342: 1309, 2000; NEJM 347: 488 & 527, 2002).
and "comet cells" (short cytoplasmic tails);
we now have PCR too (Arch. Path. Lab. Med. 123: 807, 1999;
NEJM 342: 1309, 2000; NEJM 347: 488 & 527, 2002).
|
|
|
|
CHRONIC INTERSTITIAL NEPHRITIS (CIN) is scarring of the kidney from some process that is primarily interstitial (i.e., not some glomerular or vascular disease).
{25331} interstitial nephritis
|
|
|
Once, these were all called "chronic pyelonephritis", and one important cause is of CIN is still chronic pyelonephritis.
Lupus (peritubular immune deposits) and Sjogren's syndrome (Medicine 79: 241, 2000) often include an acute or chronic interstitial nephritis; it is usually a minor problem. So does AIDS (FSGS plus round-cell interstitial nephritis suggests AIDS): Am. J. Med. 97: 145, 1994.
|
|
"Granulomatous CIN" is due to drugs or sarcoid; the latter is often severe (Arch. Path. Lab. Med. 114: 488, 1990). Acute fatianr: Nat. Clin. Parct. Neph. 4: 110, 2008.
Anti-tubular basement membrane antibody is a research tool, but its importance in human disease is not clear.
|
|
TUBULOINTERSTITIAL NEPHRITIS CAUSED BY DRUGS AND POISONS: "All drugs are poisons, and all poisons are (potential) drugs." There are several important patterns. All about drug-induced renal toxicity: Med. Clin. N.A. 74: 909, 1990.
ACUTE DRUG-INDUCED ("HYPERSENSITIVITY") NEPHRITIS
Within a month after drug exposure, the victim develops fever, skin rash, eosinophilia, hematuria, proteinuria, sterile pyuria, and/or eosinophiliuria. Withdrawal of drugs makes things better.
Methicillin is the best-known offending drug.
Sulfa drugs, rifampin (Am. J. Kid. Dis. 32: 533, 1998), cyclosporine, * penicillin, * furosemide ("Lasix"), * thiazides, and * another "Chinese herbal remedy" (Lancet 341: 387, 1993; also a carcinogen, it has ruined many, many kidneys NEJM 342: 1686, 2000) are other important causes. In fact, even in China, their "herbal remedies" are (along with antibiotics) their main cause of acute interstitial nephritis that can last a long time (Am. J. Med. Sci. 343: 36, 2012).
If biopsied, edema is always present, and interstitial mononuclear cell infiltration, eosinophils and neutrophils may be found too.
The mechanisms are obscure. Obviously, some of the problem is immunity.
NOTE: The other important cause of eosinophiliuria is atheroembolization..
ANALGESIC NEPHRITIS ("analgesic abuse nephropathy"): update (especially for radiologists) J. Am. Soc. Neph. 17: 1472, 2006
Chronic renal failure used to be extremely common in people who took phenacetin-containing combinations for pain. Phenacetin gives some people a buzz and there was a tendency to abuse it.
Aspirin, fenprofen, naproxen, phenylbutazone, indomethacin, and ibuprofen have all produced similar lesions (Tox. Path. 30: 672, 2002).
Chronic interstitial inflammation is characteristic, and many patients develop papillary necrosis (which is still very-much with us, despite the ban on phenacetin).
* Urothelial carcinoma of pelvis may also result from patients who overdid phenacetin. Around 10% of abusers develop it here or in the bladder.
CLASSIC NON-STEROIDAL ANTI-IMFLAMMATORY AGENTS (NEJM 332: 1514, 1515, 1995; Am. J. Kid. Dis. 32: 351, 1998)
This very useful class of drugs (arthritis, menstrual cramps, etc.) is also today's most common cause of renal shutdown in outpatients. The prognosis is generally excellent when the drug is withdrawn. See Ann. Int. Med. 150: 268, 1990.
When the circulating plasma volume becomes low for any reason, some people's renal microcirculation is kept open only by the local prostaglandins (PGE2 and PGI2) (NEJM 332: 647, 1995). When these people take a classic NSAID, they get vasomotor nephropathy / acute tubular necrosis, and recover in a few days. (ACE inhibitors, which prevent formation of angiotensin II, do the same thing.) Many (if not most) very-old folks given classic NSAIDs get azotemic (JAMA 264: 471, 1990; Br. Med. J. 311: 392, 1995; it happens often even today to kids in the hospital Arch. Dis. Child. 92: 524, 2007); as a matter of fact, don't give NSAID's to folks who depend on their kidneys to excrete drugs with low therapeutic indices (notably lithium).
Another typical syndrome is a combination of minimal-change glomerulopathy ("foot process disease") histology with nephritic and/or nephrotic syndrome, plus either marked chronic interstitial nephritis or acute tubular necrosis. This is infamous. Fenprofen was once considered the worst drug for causing this, but they all do it.
* Not surprisingly, if you give an NSAID alongwith a diuretic, ACE inhibitor, &/or an angiotensin receptor blocker, the risk of acute renal injury is even higher (BMJ 346: e8525, 2013).
The selective cyclooxygenase-2 (COX-2) inhibitors are much safer for the kidney (Am. J. Med. Sci. 321: 181, 2001).
LITHIUM NEPHROPATHY: mostly nephrogenic diabetes insipidus, supposedly due to fibrosis around the collecting duct.
* There may also be FSGS and cystic transformation of the collecting ducts (Lancet 376: 1024, 2010).
CYCLOSPORINE NEPHROPATHY / CALCINEURIN INHIBITOR-INDUCED NEPHROTOXICITY (becasue tacrolimus can do it also) presents lesions that, in combination, are distinctive. Big review Nat. Clin. Pract. Nephro. 2: 398, 2006.
* The acute toxicity features vacuolization of the tubules and thrombotic microangiopathy.
The chronic interstitial fibrosis that it produces seems to result from direct toxicity to the tubular epithelial cells (Am. J. Path. 167: 395, 2005).
* As the costimulation blockers like belatacept perhaps replace calcineurin inhibitors and glucocorticoids, look for better transplant function and survival, and better and longer lives for recipients. There may be more episodes of acute rejection, but less overall side-effects, including the tendency of older regimens to adversely affect serum lipids.
|
|
|
|
|
|
![]() Hantavirus in the kidney
Hantavirus in the kidney
Korean hemorrhagic fever
Yutaka Tsutsumi MD
Other poisons: lead, cadmium (Arch. Env. Health. 45: 35, 1990 for the Japanese pollution nightmare) and bismuth (interstitial nephritis, acid-fast "Cowdry A" inclusions in the proximal tubules, Fanconi syndrome), carbon tetrachloride (fatty change), etc.
HYPERCALCEMIC NEPHROPATHY (Am. J. Kid. Dis. 19: 604, 1992)
Extensive metastatic calcification of the kidney tubules (nephrocalcinosis) can cause chronic inflammation or obstruction. Because the ascending limb of Henle's loop is damaged first, one early problem is inability to concentrate urine.
Even in the absence of tubular calcification, hypercalcemia produces a prerenal azotemia by causing constriction of the small renal arteries.
A related problem (PHOSPHATE NEPHROPATHY) is acute renal shutdown in patients who may seem to have normal kidneys but who get kidney failure from calcium phosphate desposited in their kidneys after sodium phosphate bowel-preps (Arch. Path. Lab. Med. 130: 101, 2006; also NEJM 349: 1006, 2003 and J. Am. Soc. Nephr. 16: 3389, 2005). Animal model Am. J. Path. 178: 1999, 2011.
HYPOKALEMIC NEPHROPATHY
When serum potassium is very low, the kidney cannot concentrate urine.
* At autopsy, the pathologist finds coarse vacuolization of tubular cells, and dilatation of intercellular spaces (i.e., the tubular cells look white and broken).
PLASMA CELL MYELOMA KIDNEY (Arch. Path. Lab. Med. 128: 875, 2004; Blood 116: 1397, 2010)
{17274} plasma cell myeloma, cast; PAS stain shows magenta tubules
In "cast nephropathy", the problem is precipitation of Bence-Jones protein within the distal tubules causing renal shutdown. Patients experience the acute or insidious onset of renal failure.
The pathologist will see amorphous pink cast-plugs, often surrounded by a foreign-body reaction with multinucleated giant cells. There's a considerable range of pathology, and the proximal tubular cells may contain curious findings as well: Arch. Path. Lab. Med. 131: 1368, 2007.
* Molecular biology and physics of how the casts do the damage: J. Clin. Inv. 122: 1777, 2012.
* If you induce dehydration or osmotic diuresis in a plasma cell myeloma kidney patient (as, by ordering an intravenous pyelogram), you will destroy their kidneys.
* Light-chain nephropathy can also give the same tubular casts.
Remember that plasma cell myeloma patients often get amyloidosis AL.
OXALATE NEPHROPATHY
Antifreeze drinkers (ethylene glycol), inborn errors (Am. J. Kid. Dis.
19: 546, 1992), extreme ascorbic acid abusers (Arch. Int. Med. 950, 1985), and * those infected with
"Aspergillus niger"![]() ,
which produces oxalate as its waste product (Arch. Path. Lab. Med. 119: 558,
1995).
,
which produces oxalate as its waste product (Arch. Path. Lab. Med. 119: 558,
1995).
* The anesthetic gas methoxyflurane was banned for causing oxalate nephropathy.
* The thankfully-rare inborn error of metabolism "primary hyperoxaluria" ruins kidneys and livers.
Sharp oxalic acid crystals ruin the kidney. Long suspected, it's now proved that it is indeed the crystals that do the damage in ethylene glycol poisoning (Clin. Tox. 47: 859, 2009).
RADIATION NEPHRITIS
Following therapeutic radiation involving the kidney, the interlobular arteries narrow. This eventually causes high blood pressure and sometimes renal failure.
* Not surprisingly, children given total-body radiation to cure their leukemia sometimes end up with renal failure (Arch. Dis. Child. 72: 382, 1995).
* BALKAN NEPHROPATHY (update Am. J. Neph. 26: 1, 2006; Kid. Int. 69: 644, 2006)
A rapidly-progressive interstitial nephritis that occurs in about ten, very sharply-defined areas of Rumania, the former Yugoslavia, and Bulgaria. Epidemiologically, the disease typically occurs in farm workers (and their pigs: Vet. Rec. 142: 190, 1998), and shows a strong familial tendency ("Danubian endemic familial nephropathy"). Obviously some toxin in the environment is interacting with genes.
The pathology is that of tubular loss, interstitial fibrosis, and myxoid thickening of the intima of small arteries. The subcapsular cortex is most severely involved. Renal failure develops in a few months or years. There are three competing ideas about the cause.
1. The fungal poison OCHRATOXIN A was abundant in moldy grain stored in the upper stories of houses. The particular school of communism in the Balkans opposed barns for ideological reasons. Supporting the link to a fungal toxin: These patients, and their neighbors, also often develop urothelial cancer, and ochratoxin is a urothelial carcinogen. Of course there may have been other factors operating, but the geography of the fungus and the disease matched closely. There's a nearly-identical fungal nephropathy in Tunisia: Arch. Tox. 69: 553, 1995; Tox. Lett. 83: 869, 1995.
2. An illness very similar to Balkan nephropathy was produced when ARISTOLOCHIA, a poisonous plant, ended up in a "holistic Chinese herbal remedy" for obesity, prompting an epidemic of kidney failure and urothelial cancer: NEJM 342: 1686, 2000. The similarity of the Balkan nephropathy lesion to the aristolochia fiasco prompted an study and, sure enough, people in the endemic regions remembered Aristolochia clematitis (European birthwort) growing in the wheatfields (Croat. Med. J. 46: 116, 2005).
3. Since the disease did not disappear with the improved living conditions after the fall of communism, perhaps the cause is toxins from the low-rank coals leaching into the well water on the involved farms. For now, I'm betting it is the Aristolochia.
* RENAL HYPERTROPHY
The cells of the proximal and distal convoluted tubules (and, to a lesser extent, the other cells of the nephron) have a limited ability to increase in size.
This occurs in a solitary kidney (congenital, after unilateral nephrectomy), and sometimes in chronic metabolic derangements.
NEPHROGENIC DIABETES INSIPIDUS
A family of diseases in which the collecting duct is unable to respond to ADH. This includes inborn errors of metabolism (Nature 359: 233, 1992; lack of ADH / V2 receptor, * aquaporin or other proteins in its let-the-water-back-out cascade), gross and widespread damage to the medulla, and the late effects of lithium therapy for mania.
GOUT: An Important Kidney Disease. Update Lancet 375: 318, 2010; JAMA 308: 2133, 2013.
{25521} gout, tophi
{25522} gout, tophi
{24641} gout, bone
{11559} gout, histology of tophi; uric acid crystals
are pale and surrounded by granuloma
{24642} gout, uric acid crystals under polarized light
{24643} gout, uric acid crystals unpolarized; they are brown
|
|
|
|
Australian Pathology Museum High-tech gross photos
|
|
Uric Acid Metabolism: Uric acid is the catabolic end-product of purines from food and from broken-down cells. It is excreted in the urine. (The way in which the kidney handles it is interesting.) The complexities of normal uric acid metabolism have been worked out by biochemists. The mysteries of how it is transported in our bodies have been more elusive, and there's now an extensive literature (J. Clin. Inv. 120: 1791, 2010). Gout is due mostly to abundant purines in the diet and problems getting it out of the body. It is becoming more common as people are eating more meat.
Introduction and Classification: Gout is the disease resulting from precipitation of monosodium urate crystals in the body. Everyone knows the classic story. Gout strikes the middle-aged, overweight man, who after an evening of good food and fine wine, is awakened at 2 AM by an agonizing pain and swelling in the first joint of one of his big toes. The acute attack lasts several days and ends as mysteriously as it began, perhaps returning on another occasion when the patient has "indulged".
All patients with too-high serum uric acid (i.e., urate) levels from any cause are said to have hyperuricemia. (Hyperuricemia is an abnormal lab test, gout is a disease.) Only hyperuricemic patients can get gout, but most never do. Up to 15% of adult men have serum uric acid levels in excess of uric acid's theoretical solubility (7 mg/dL), but perhaps one adult man in 200 has an attack of gout per year. There is surprisingly little correlation between serum uric acid elevation and the presence and severity of the gout.
Gout patients are classified according to their current symptoms and signs. The typical patient with gout has repeated, transient, severe inflammation of one or a few joints (most commonly the coldest ones, i.e., the big toe). This is "acute gouty arthritis". Longstanding gout results in deposition of crystals of monosodium urate surrounded by a chronic inflammatory reaction with a foreign body reaction and fibrosis. The whole thing is called a "tophus" (plural "tophi", and yes, they do count as granulomas.) Tophi are common in the helices of the ears, in the joints and their bursae ("chronic tophaceous arthritis"), and in the kidneys. (Feel the finger pads for tophi before gouty arthritis manifests: Arch. Int. Med. 148: 1830, 1988.) Occasionally a tophus can weaken a bone sufficiently to cause a pathologic fracture (kneecap Clin. Orth. 445: 250, 2006). Laryngeal tophi can compromise the airway (J. Laryngol. Otol. 116: 140, 2002). Sometimes gout presents not with joint problems, but as a uric acid kidney stone or some other manifestation of "renal gout".
Gout patients are also subdivided by etiology. Primary gout is any form of gout caused by a known or assumed inborn error of uric acid metabolism or transport, without other systemic manifestations. It includes primary idiopathic gout (the great majority of gout sufferers), and a few rare, well-characterized genetic syndromes. Secondary gout is hyperuricemia and resulting gout because of some other disease. These are outlined below. But the tissue changes are the same in primary and secondary gout.
Etiologies of gout: Primary Hyperuricemia and Gout (90+%) ("idiopathic gout", "common gout") In most cases, the reason for the hyperuricemia is unknown (and assumed to be "genetic and multifactorial"). While the condition runs in families, the molecular biology has eluded scientists, various mechanisms are at work in various patients, and there is no simple pattern of inheritance. The majority of these men both overproduce and underexcrete uric acid. Some cases are less idiopathic than others. Several "partial enzyme defects" (* Including an overactive phosphoribosyl pyrophosphate synthetase) have been identified. Primary idiopathic gout is a disease of adult males (95%). Attacks peak during the 40's, though the hyperuricemia begins at puberty.
Secondary Hyperuricemia and Gout (5-10%): Hyperuricemia is common in certain conditions, but actual gout is uncommon. Heavy ingestion of purines (i.e., organ meats) can raise serum uric acid and trigger an acute attack of gout. Diseases with increased cell turnover or widespread cell destruction increase uric acid production. Remember lymphoma, leukemia, other malignant tumors, sometimes even psoriasis (in which the epidermal cells undergo extremely rapid proliferation). Cancer patients receiving chemotherapy and/or radiation that works often excrete tremendous amounts of uric acid. People with functional obesity have increased production and diminished secretion of uric acid. The reasons are unknown. In 2008, somebody noticed that fructose (from corn syrup in soft drinks) raises uric acid and correlates with gout (BMJ 336: 285, 2008). In diseases of the proximal tubule (most notably lead poisoning), secretion of uric acid is poor and gout can result. Secretion is also inhibited by thiazide diuretics, ethanol, and acetoacetic acid. (Only about 5% of patients with chronic kidney failure get clinical gout.) Uric acid reabsorption is increased in low-volume states with slow flow through the proximal tubules, and in heavy alcohol use.
If gout is part of a systemic disease with other problems, it's called "secondary", whether it's genetic or not. Two genetic syndromes in which gout is one of many problems are....
Type I Glycogen Storage Disease (von Gierke): Gout in this disease result from over-production of uric acid plus and decreased urate secretion. These are due to the presence of abnormal carbohydrate metabolites.
Lesch-Nyhan Syndrome: An X-linked recessive trait with complete deficiency of "salvage enzyme" (hypoxanthine-guanine phosphoribosyl transferase, HGPRT). Therefore, hypoxanthine cannot be salvaged, more hypoxanthine becomes uric acid, and more purines must be made from scratch (only to become more uric acid). Severe gout is one feature of this terrible disease. A less complete deficiency is one known cause of "primary gout".
The Acute Gouty Attack: The attacks begin and end so suddenly, and are so severe, that a vicious cycle obviously underlies them. It is preceded by precipitation of insoluble monosodium urate crystal in relatively avascular tissue (cartilage, epiphyseal bone, periarticular bone, extraskeletal). This has probably been going on during the years of hyperuricemia, but sequestered somehow without producing symptoms. The acute attack begins when some crystals effectively activate the plasma proteases. Monosodium urate crystals themselves can activate factor XII and C5. Monosodium urate crystals can also adsorb whatever opsonins may be around, rendering them tasty to phagocytes. Polys and monos soon arrive and start trying to eat the crystals. The crystals then rupture the lysosomes and lyse the phagocytes. Enzymes from the dead phagocytes enter the synovial fluid and damage tissue, activate complement, etc. More phagocytes arrive and the same thing happens again. Whatever urate crystals may be present in tophi or microtophi within the joint will probably be released as the process continues. Plus, as the inflammation causes the pH in the joint to go down, more and more urate crystallizes. Acute attacks probably are self-limited because the heat of inflammation redissolves the crystals.
The Chronic Disease: Although the acute attack is the most dramatic aspect of gout, the enduring lesion of gout is the tophus. Future pathologists -- the crystals are water soluble, so unless you process the tissue specially, they will dissolve away. (Use absolute alcohol). Future clinicians -- when you suspect gout, feel for tophi in the helix and antihelix of the pinna. Joints: Repeated acute attacks, or uric acid buildup, wrecks the cartilage and underlying bones, which may become fused, lipped, etc. The most commonly involved site is the big toe, but other cold places, such as the elbows, wrists and fingers, are often involved. Perhaps one fourth of gout patients develop uric acid kidney stones. Future radiologists -- remember that, unlike most kidney stones, uric acid stones are radiolucent. A sudden rush of uric acid at low pH within the renal tubules can shut them down ("acute urate nephropathy"). Tophi in medulla and pyramids ("chronic urate nephropathy") cause acute and chronic kidney problems. Finally, patients with hyperuricemia tend to develop high blood pressure. Why this happens remains unknown.
Clinical Diagnosis: Some reminders. (1) The patient with gout is seldom the first family member affected. (2) Serum uric acid is routine on most automated chemical profiles. (3) Compensated polarization of synovial fluid, tophus, or kidney stone can identify the uric acid crystals. (4) "Uric acid crystals" and "amorphous urates" reported on urinalysis are of essentially no significance, and are common in normals with acid urine.
Treatment: Colchicine, the mitotic spindle poison from the crocus flower, stops neutrophils from moving into the involved area (it paralyzes them) and stops the acute attack. Non-steroidal anti-inflammatory agents are also useful for the joint disease. Allopurinol, a purine analogue, inhibits xanthine oxidase and prevents the formation of uric acid. The precursors are excreted instead. Allopurinol, though expensive, is the mainstay of chronic treatment of gout. It is now getting some competition from febuxostat, a new nonpurine xanthine oxidase inhibitor (NEJM 353: 2450, 2005; passes its five-year study Rheumatology 48: 199, 2009). Allopurinol is perhaps the most infamous single drug for causing necrosis of the entire epidermis -- watch for "rashes". Probenecid makes renal clearance of uric acid more effective (helps the hyperuricemia and joint disease, but clearly not a way to prevent kidney stones from forming in over-producers). If the urine is kept alkaline, uric acid is much less likely to precipitate in the kidney.
The Johns Hopkins prospective Gout Study: JAMA 266: 3004, 1991. The fact that there has been very, very little work on gout since tells me that this disease is basically defeated.
|
|
|
HIGH BLOOD PRESSURE ("hypertension")
A long-standing increase in systolic and/or diastolic blood pressure above desirable levels, i.e., 140/90 or thereabouts). This affects maybe 15% of people in the U.S., largely (but not exclusively) among older people.
* Please review: The never-ending flap over "who needs to be treated". If you believe the WHO recommendation that normal is "systolic 130 or under", you'll believe anything (Lancet 355: 175, 2000). Reasonable people seem to agree that more than 150/90 is too high for folks over 60, most people think over 140/90 is too high for folks under 30, and that clinical judgement should rule (JAMA 311: 507, 2014).
Classification
90% idiopathic (essential hypertension)
This includes the common "benign essential hypertension" and the commonest form of the rare "malignant hypertension." (Clinically, you'll diagnose "malignant" hypertension if it causes papilledema, or maybe if blood pressure's over 200/140 and the patient is suddenly ill. At autopsy, you'll diagnose "malignant" hypertension is arterial walls are necrotic.)
10% secondary hypertension (review Hosp. Pract. 29(4): 137, 1994 April 15, 1994)
Endocrine diseases: Cushing's, 11-beta-hydroxylase deficiency and its kin (J. Clin. End. Metab. 77: 687, 1993), hyperaldosteronism, pheochromocytoma, hyperparathyroidism (high calcium constricts vessels), eclampsia (a tremendous killer of women, especially in the poor nations: Br. J. Ob. Gyn. 99: 547, 1992), reninoma, bad hypercalcemia from some other cause (unusual), licorice abuse (really, * It inhibits cortisol oxidase and 11-beta-hydroxylase: NEJM 325: 1223, 1991), Liddle's syndrome, others.
Renal hypertension: coarctation of aorta, known renal or renal arterial disease
Nobody understands where diabetes fits into the picture. Hypertensive diabetics tend to hang onto sodium for some reason.
* Nobody really knows where sleep apnea fits into all this, but the relationship (at least) is impressive independent of obesity (Chest 104: 775, 1993; Am. J. Med. 91: 190, 1991; questioned Chest 102: 367, 1992; more Am. J. Med. 98: 118, 1995, Br. Med. J. 320: 479, 2000).
Nobody understands, either, why low birth weight predicts adult hypertension, but the effect seems powerful. The effect increases depending how low the birth weight was, and how old the patient now is (Br. Med. J. 306: 24, 1993). Sounds like a positive-feedback things that starts in the womb.
* Two molecules to watch are chromogranin A, which is overabundant in essential hypertension, and its catecholamine-release-inhibiting fragment catestatin (J. Clin. Inv. 115: 1942, 2005).
Not included in this classification are the causes of increased pulse pressure, due to loss of arterial compliance (atherosclerosis, really extensive Monckeberg's) or greatly increased cardiac output (exercise, emotion, fever, hyperthyroidism, AV fistulas, Paget's of bone, beriberi, aortic valve insufficiency, patent ductus arteriosus).
Regardless of cause, longstanding high blood pressure is bad. Patients get:

BENIGN ESSENTIAL HYPERTENSION (common "high blood pressure", "the silent killer", etc.);
Affects about 15% of the population, prevalence increases with age. Five percent of people in 30's, 30% of people in 50's. All ages females more than males, blacks more than whites. A physician's long personal history of high blood pressure and its treatment over decades: Br. Med. J. 298: 445, 1989.
Usually slow, progressive, asymptomatic increase in blood pressure.
What causes "benign" essential hypertension? This continues to elude us:
The basic trouble in most patients does seem to be generalized arteriolar vasoconstriction and/or inability to dump a salt load.
Vasoconstriction by catecholamines ("released in response to emotional stress") figured prominently in older explanations, especially those by psychiatrists. The sympathetic nervous system as a factor in hypertension (Am. J. Med. Sci. 303: 271, 1994).
* The idea that hypertensives' arteries won't dilate when they should has found new support from measurements of the retinal vessels in two big prospective studies. The artioles narrow BEFORE high blood pressure appears (Br. Med. J. 329: 79, 2004; Ann. Int. Med. 140: 138, 2004) and may prove to be a cardiac risk factor (Heart 92: 1583, 2006).
Media-to-lumen ratio (i.e., just how much too thick is that wall?) seems to be the foremost predictor of disaster in hypertension, regardless of cause (Circulation 108: 2230, 2003).
The larger arteries also take a beating. Fibroelastic thickening of the intima smooth muscle hypertrophy in the media both occur in the larger and medium-sized arteries; this regresses when the hypertension is treated (Circulation 101: 2601, 2000).
Watch for sporadic rat-borne hantavirus infection as an explanation for why some people just turn bad-hypertensive. Only 0.25% of adults have antibodies against any hantavirus, but 6.5% of people with end-stage renal disease due to hypertension (but not other uremics) have such antibodies. J. Inf. Dis. 167: 614, 1993. There's a Balkan hantavirus that devastates the renal vasculature (J. Inf. Dis. 166: 113, 1992), and the Korean one is old news (Ann. Int. Med. 113: 385, 1990). Currently, hantavirus is a "usual suspect" in the US for "mysterious" ATN, "mysterious" IgA nephropathy, and "mysterious" segmental necrotizing glomerulonephritis (Am. J. Kid. Dis. 33: 734, 1999).
The molecular biology is only starting to be understood. Whatever is really happening, the kidneys of most hypertensives seem to have difficulty disposing of a sodium load. (And, of course, we in the US eat too much salt.... The Gates Foundation people's estimates of 1.65 million excess deaths each year from excess sodium consumption seems way-low to this pathologist NEJM 371: 624, 2014). However, salt-loading will not cause high blood pressure in most people.
The American salt craving: Am. Sci. 75: 27, 1987. "Salt abuse: the path to hypertension": Nat. Med. 14: 16, 2008 -- focuses on lasting changes in vascular myosin phosphorylation caused by longstanding salt-overloading (Nat. Med. 14: 64, 2008). The pop notion that everyone should avoid salt has been called both unscientific and probably false (Lancet 351: 781, 1998) -- however, if the new work is accurate, we may want to reconsider. Interestingly, no one looks at how much water folks drink... which we'd expect to remove salt from the system. Stay tuned; this will get more confusing. Salt intake reduction as a public health education initiative: NEJM 362: 650, 2010.
One idea, still popular, involves diminished production of nitric oxide ("endothelially-derived relaxation factor", "EDRF"; NEJM 323: 22, 1990; cause or result?). Now we know that the production of this stuff falls off as we get old (J. Geront. 49: B-157, 1994).
* Didja know that sodium nitroprusside works by producing nitric oxide?
* Some other suggestions focus on other mediators, such as prostaglandins, lack of atrial peptides, etc., etc,. A mouse with deficient atrial natriuretic peptide has sodium-sensitive hypertension (Science 267: 679, 1995), just as you would expect.
* Hypertensives seem to have increased activity of Na+-H+ exchangers on a variety of cell membranes. There is talk of increased calcium channels, etc., etc.
It's well-known that hypertensives' small arterioles eventually lose the ability to dilate appropriately. This is probably due at least partly to the hyaline (etc.) arteriolar sclerosis that develops from the hypertension.
Endothelin is often elevated in hypertension, regardless of etiology. At the very least, this seems to perpetuate the problems. Endothelin supposedly constricts the renal vasculature, makes the mesangial cells contract, prevents resorption of sodium and water, and causes hyaline to accumulate in vessels and gloms. Since the kidney must clear it, enhanced levels of endothelin and renal failure probably promote each other. In 1994, I predicted endothelin would prove a major player; it is now (with insulin) the "primary suspect" in the hypertension that tneds to run with the metabolic syndrome (J. Clin. Endo. Metab. 92: 379, 2007).-
* Mouse model in which superoxide radicals destroy EDRF and cause hypertension: Proc. Nat. Acad. Sci. 88: 10045, 1991. All about endothelin: NEJM 333: 356, 1995.
Much work in common high blood pressure still involves angiotensin II.
* The good allele is "I", the bad one "D". Angiotensin II is an important growth factor, and it's now considered the main cause of fibrointimal hyperplasia and cardiac myocyte hypertrophy (Circulation 101: 148, 2000).
Hereditary factors (the hypertension genes: Science 272: 676 1996 is still good, as others have been elusive), personality variables, environmental stress (for example, job strain: JAMA 263: 1929, 1990; West. J. Med. 153: 180, 1990; and of course the classic "repressing your anger", which has found new support JNMD 177: 15, 1989), and obesity (Ann. Int. Med. 128: 81, 1998) all seem important in hypertension.
Obese patients, and black patients who are developing hypertensive renal insufficiency, are likely to have much increased plasma volume (i.e., total-body sodium).
Among blacks, both genetics (there must have been some selection for a tendency to retain salt under the deplorable conditions of slavery) and environment (salt in the diet, socioeconomic stress) appear to be important (JAMA 265: 599, 1991; Am. J. Med. Sci. 307(S1): S-38, 1994; Am. J. Med. Sci. 304: 306, 1992); barriers to care also exist (Am. J. Kid. Dis. 19: 397, 1992).
Update on the especially aggressive nature of hypertension in blacks: Arch. Int. Med. 168: 832, 2008.
Young adults with newly-discovered high blood pressure are likely to have inappropriately high cardiac output with normal peripheral resistance (i.e., they do not have generalized vasoconstriction), usually are not sodium-overloaded, etc.
The benefits of exercise (aerobic, strength training) in the treatment of hypertension seem less impressive than the common wisdom suggests: JAMA 266: 2098, 1991. A realist writes on diet in the pathogenesis and treatment of hypertension (Med. Clin. N.A. 77: 831, 1993; "we don't know much about causes, we can't promise much benefit, we can't expect compliance").
You already remember the problems caused by hypertension on the microvasculature. Stiffening of the arteries and arterioles has long been measurable, and now researchers are actually measuring the numbers of circulating endothelial cells (i.e., cells that were forced off the inner walls by hypertensive damage -- larger numbers indicate worse outcomes (Chest 132: 1540, 2007; J. Clin. Endo. Metab. 96: 3175, 2011).
Whether cutting the sympathetic nerves that supply the kidneys will help medicaton-refractory hypertension remains to be seen. A novel new intervention is creating an artificial arterio-venous fistula between the extenal iliac artery and vein (Lancet 385: 1596 &1634, 2015; I expect this will work but will throw many people into high-output heart failure.)
ARTERIOLAR NEPHROSCLEROSIS ("benign nephrosclerosis")
Seen in patients with high blood pressure from any cause; thus, the kidney lesion of "benign essential hypertension", the commonest cause.
Smallish, symmetric kidneys with finely granular (sandpaper) surfaces. (Each pit is the site where a nephron has been lost and the scar has contracted.)
Some glomeruli are replaced by collagen, with corresponding tubular atrophy and interstitial fibrosis. If you do a PAS stain, you'll see the remnant of the tuft collapsed near the pole and encased by collagen.
The small renal arteries and afferent arterioles of hypertensives show a variety of changes:
{21010} arteriolar nephrosclerosis with plenty of little cysts (gross)
{10575} arteriolar nephrosclerosis, sandpaper kidney
{11777} arteriolar nephrosclerosis, hyalinized artery
{40267} arteriolar nephrosclerosis, gross
{25760} hypertension, bad fibrous intimal proliferation
{08871} hypertension, intimal fibroplasia
{25764} hypertension, intimal fibroplasia
|
|
All these changes are even more pronounced in diabetics, though diabetes also hyalinizes the efferent arterioles. Treating hypertension in diabetics, especially young ones, is a must: Ped. Clin. N.A. 40: 81, 1993.
{25763} hypertension
{25764} hypertension
{25765} hypertension; trichrome
{25766} hypertension; reticulin
Some such arteriolar changes are seen in any elderly person, or anyone with high blood pressure affecting the kidney, from any cause. It starts in young people: Arch. Path. Lab. Med. 116: 50, 1992. It seems an inevitable accompaniment of aging (Ann. INt. Med. 2010: 152, 561, 2010).
When arteriolar narrowing develops, it may contribute to the hypertension, but is not the primary cause.
Uncomplicated arteriolar nephrosclerosis causes renal failure in less than 5% of its victims (mostly blacks).
However, you want to prevent arteriolar nephrosclerosis. This is one of the reasons you want to treat hypertension early (Arch. Int. Med. 150: 2073, 1990).
"Bright's riddle": Dr. Bright noticed that patients at autopsy who had left ventricular hypertrophy often had small kidneys with finely-granular surfaces, and vice versa.
Perhaps not surprisingly, one study found about 50% of "hypertensive" children merely have white jacket hypertension (J. Fam. Pract. 33(6): 617, Dec. 1991).
MALIGNANT HYPERTENSION: High blood pressure causing papilledema (clinically) or necrosis of the vessels themselves (autopsy)
Hypertension from any cause can accelerate or enter a malignant phase.
Malignant hypertension may develop in previously healthy patients, but usually is superimposed with pre-existing benign hypertension (develops in 1% of cases).
It is more common in males than females; more common in blacks than Caucasians.
Scleroderma and adult hemolytic-uremic syndrome often terminate in malignant hypertension. (All three display onion-skinning of the intima of the small renal arteries.) Scleroderma kidney Kid. Int. 41: 462, 1992.
Cocaine abuse precipitating malignant hypertension: Am. J. Med. Sci. 312: 295, 1996. Now all-too-well-known: Arch. Path. Lab. Med. 131: 1817, 2007.
{25757} malignant hypertension, bleeding into the kidney
{25759} malignant hypertension, most of kidney is infarcted
{25761} malignant hypertension, intimal proliferation (trust me this was malignant hypertension)
{25762} malignant hypertension, fibrinoid necrosis
{25762} malignant hypertension
|
|
|
|
|
Patients have rapidly rising blood pressure, generally reaching levels greater than 200/110. This has many bad effects.
Hypertensive encephalopathy (papilledema, behavioral changes, nausea and vomiting, headache, visual disturbances, seizures)
Congestive heart failure (ask a cardiac pathophysiologist; increased volume or resistance must be rough on the heart) or intracerebral hemorrhage
Malignant nephrosclerosis: the renal lesion of malignant hypertension
The essential feature is damaged, non-inflamed arterioles.
Glomeruli show scattered necrosis and hemorrhage. Bleeding into the glomerular spaces appear as petechiae on the cortical surfaces ("flea-bitten kidneys", seen also in the nephritic syndrome).
Intimal proliferation ("onion-skinning") of small arteries is characteristic; this badly narrows the lumens, renin levels increase, and renal infarcts occur.
The media fills with "fibrinoid" because of endothelial damage. (There may also be some necrosis of the media.)
Of course, the kidneys will also show "arteriolar nephrosclerosis" if there was pre-existing "benign" hypertension.
Malignant nephrosclerosis destroys the kidney in a few months (except in a minority of patients who die of stroke).
RENAL VASCULAR HYPERTENSION:
Renal artery stenosis
It is usually caused by atherosclerosis of an ostium, in older folks.
Adult polycystic kidney plays havoc with the microvasculature, and high-renin hypertension occurs early (NEJM 323: 1091, 1990).
Fibromuscular dysplasia (actually a congenital malformation, and not anaplastic or pre-malignant) is a rare cause of this in young people. The beaded appearance of the renal artery on angiography is typical. Treating it with an endovascular prosthesis: NEJM 336: 459, 1997.
* It's fairly common in neurofibromatosis type I.
* Takayasu's arteritis, * von Recklinghausen's neurofibromatosis, and * adhesions from previous surgery are the other causes.
"Goldblatt's riddle": Dr. Goldblatt noticed that many patients at autopsy who had high blood pressure in life possessed one normal-sized kidney and one small kidney with a narrowed renal artery (the "Goldblatt kidney").
Dr. Goldblatt answered his own riddle by producing high blood pressure in animals, using a clamp to produce subtotal occlusion of one renal artery.
"Goldblatt hypertension" exhibits (at least at first) high serum renin levels. (* Stay tuned for the introduction of renin inhibitors into clinical practice: Nature 357: 446, 1992).
The kidney supplied by the narrowed artery is often small and fibrotic and may contain infarcts from atheroembolic episodes.
* Under the microscope, the kidney downstream from the narrowing is likely to have normal glomeruli and ischemically atrophied tubules.
Eventually, however, the resultant hypertension will cause the other kidney to show changes of nephrosclerosis. The kidney with the renal artery stenosis will show less arteriolar change than the other kidney, due to the protective effect of the stenosis. (So the "Goldblatt kidney" is sometimes the larger one.)
Another tipoff is that these patients tend to have good responses to angiotensin converting enzyme (ACE) inhibitors (why?) In cases where there is a birth defect in an artery, this is likely to be a surgical or angioplasty disease.
* Stenting of an atherosclerotic renal artery seems to give little benefit over good general care: NEJM 370: 13, 2014.
* "Low blood pressure", a fad diagnosis in France and Germany, is probably a myth (Br. Med. J. 301: 362, 1990).
HEMOLYTIC-UREMIC SYNDROME ("HUS"; review of the thrombopathy NEJM 346: 23, 2002):
A pattern of renal injury with several causes, all with platelet clumping in small renal blood vessels, resulting in microangiopathic hemolysis, thrombocytopenia, and acute renal failure. (Don't confuse HUS with DIC, as the clotting factors are not consumed with the platelets; fibrinogen levels, PT, and PTT all stay normal.)
The pathogenesis is just now being worked out. The fundamental problem is selective damage to the endothelium of the renal microvasculature.
* As you'd expect, there are a few known genes (for example, a C3 convertase mutation J. Immuno 188: 2030, 2012; a family of six others Blood 119: 591, 2012); all are late-onset because if they affected the child before birth, they would result in miscarriage.
Childhood hemolytic-uremic syndrome (NEJM 323: 1050 & 1161, 1990) is an important epidemic disease, presently becoming more frequent, and is now the most important cause of acute renal shutdown in kids -- most of whom recover, but around 10% have residual damage.
It usually follows viral illness or bacterial bloody diarrhea (* "D+ HUS"). The child usually recovers completely (* "D- HUS" is more mysterious and more ominous).
There were epidemics in California in the early 1980's. The epidemic in Minnesota: NEJM 323: 1050 & 1167, 1990. Remember the "Jack-in-the-Box" hamburger fiasco (JAMA 272: 1349, 1994)? The sprouts outbreak in Germany was horrorific: NEJM 365: 709 & 1835, 2011; brain damage was part of the syndrome Lancet 378: 1166, 2011. The source in Germany was sprouts from one "organic" farm, the microbe an unusual E. coli that had acquired the ability to make Shiga toxin 2 and be resistant to antibiotics. Petting-zoo HUS: MMWR 54: 1277, 2005.
The best-established cause (* of "D+") is verocytotoxin ("shiga toxin"; "Shiga-like toxin", Stx), a poison from certain strains of E. coli (notably 0157:H7, which spread in the 1990's cattle; especially beware of rare burgers (the bugs can survive on the inside and are likely to be there because the meat is ground) and raw milk for kids -- Postgrad. Med. 88: 135, Oct. 1990; Br. Med. J. 303: 800, 1991; Lancet 337: 138, 1991; NEJM 333: 364, 1995). The related "Shiga toxin" from Shigella dysenteriae does the same thing. This damages endothelial cells, and causes them to release factor VIII aggregates, which clump platelets (Nephron 51: 207, 1989).
Once the process gets going, neutrophil products probably contribute to the general mayhem.
* Future blood bankers! Verocytotoxin, which binds to specific receptors on the renal endothelial surfaces, also binds to P1 red cell antigen, which buffers it. Kids lacking this antigen are at much greater risk for HUS.
"Atypical hemolytic-uremic syndrome", with a poor prognosis and not always a clear cause, occurs about half the time in folks with various mutations in complement modulating proteins; an additional 5% have mutant thrombomodulin (THBD; NEJM 361: 468, 2009). Update on "atypical HUS": NEJM 1676: 361, 2009. Treating it with eculizumab, the anti-complement monoclonal antibody: NEJM 368: 2169, 2013.
One to watch: Hemolytic-uremic syndrome histology is now a recognized complication of treatment with bevacizumab, the anti-VEGF cancer treatment (why might this be?). See NEJM 358: 1129, 2008; Am. J. Med. 122: 322, 2009; BMJ 344: e3838, 2012.
ADULT HEMOLYTIC-UREMIC SYNDROME:
This follows complications of pregnancy (* rarely exposure to exogenous estrogens), administration of endothelial poisons, notably * mitomycin C (an anti-cancer drug) * cyclosporine, * "collagen- vascular diseases", and * crack cocaine (Am. J. Med. Sci. 299: 366, 1990). It typically ends in chronic renal failure and sometimes malignant hypertension.
Biopsy shows intimal onion-skinning in small renal arterioles (* the more of this, the worse the prognosis), fibrin thrombi and/or red cell fragments in the glomeruli, and "fuzz" (a mix of fibrin and amorphous junk) in the subendothelial spaces.
THROMBOTIC THROMBOCYTOPENIC PURPURA has only superficial similarities to HUS. The thrombi are widespread (not just confined to the kidneys as in HUS), mostly composed of platelets, neurologic defects are more prominent, and the patient is either cured or dies.
THROMBOTIC MICROANGIOPATHY is now an umbrella term for a host of renal lesions that have in common damage to the endothelium.
THE FINDINGS IN THROMBOTIC MICROANGIOPATHY
DIFFUSE CORTICAL NECROSIS
{10570} diffuse cortical necrosis
{10574} diffuse cortical necrosis
{21026} diffuse cortical necrosis
The terminology is being revised, but this is necrosis of the cortex that isn't going to get better (i.e., the stroma is also destroyed, unlike in "acute tubular necrosis"). This usuallly results from severe DIC plus shock, usually post-partum. (* Snake bite: Kid. Int. 35: 891, 1989). Unlike in HUS, the kidney vessels are normal.
The patient, of course, has acute renal failure that doesn't get better. At autopsy, fibrin thrombi plug the glomerular capillaries. (They appear here first, of course, because protein is concentrated here.)
ATHEROEMBOLIZATION (Lancet 375: 1650, 2010)
{16900} atheroembolus; scar has formed around it
|
|
A common cause of acute renal failure in older patients, especially those with abdominal aortic aneurysms.
The new thrombolytic therapies for myocardial infarctions tend to remove surface thrombi from ulcerated atherosclerotic plaques, releasing their contents. You'll see a few kidneys destroyed in this way.
The diagnosis is difficult to make during life. (* It doesn't really matter, because atheroembolization isn't treatable -- one group after reviewing the difficulty of making the call found that statins help some, even after the event: Circulation 116: 298, 2007). Look for livido reticularis on the lower extremities, increased eosinophils in the blood and urine.
At autopsy, the pathologist finds platelike ("needle-shaped") crystals of cholesterol in the renal arteries, with an associated foreign-body reaction.
Survivors of this (or any other sort of renal embolization) end up with a kidney with one or more V-shaped scars ("arterial nephrosclerosis"). You will see V-shaped scars (i.e., narrow and deep) whenever the distribution of entire interlobular arteries is destroyed (and replaced by contracted scar).
SICKLE CELL DISEASE NEPHROPATHY (South. Med. J. 104: 752, 2011)
Cells tend to sickle in the hypertonic milieu of the renal medulla, even in heterozygotes. This result in hematuria and/or decreased ability to concentrate urine, loss of vasa recta, renal tubular acidosis (type 4), etc.
Sicklers (HbSS, HbSC) often get papillary necrosis (even carriers' cells sickle in the hypertonic milieu), many other problems.
* The glomeruli are affected in about 50% of sickle cell disease patients as well. There's a roughly equal division among focal-segmental glomerulosclerosis, membranoproliferative type I, FSGS, and simply clogged capillaries (Medicine 89: 18, 2010).
Renal medullary carcinoma is a rare cancer seen almost exclusively in people with sickle cell disease or trait. You won't be asked to recognize it, but it's a good thing to know on rotations. Update Am. J. Surg. Path. 37: 368, 2013.
OBSTRUCTIVE DISEASE OF THE KIDNEY
Causes include malformations, blood clots, calculi, tumors, prostatic hyperplasia, * retroperitoneal fibrosis, and neurologic bladder problems.
Clinical pictures:
Unilateral obstruction may be clinically silent.
Bilateral partial obstruction will first manifest as loss of concentrating ability due to tubular atrophy.
Bilateral complete obstruction results in anuria.
Obstruction produces dilatation of the calyceal system (hydronephrosis), eventually with fibrosis and tubular atrophy of the kidney (relative sparing of glomeruli. Of course, there are many other causes of hydronephrosis).
Obstruction predisposes to acute and chronic pyelonephritis and (less often) renal papillary necrosis.
{10573} hydronephrosis
{10577} hydronephrosis
KIDNEY STONES (renal calculi, "urolithiasis"; Am. Fam. Phys. 63: 1329, 2001; BMJ 334: 468, 2007)
{40264} kidney stone
{09786} staghorn kidney stone
{09789} kidney stone
{18787} staghorn stone
{49322} kidney stone
|
|
Kidney stones form in the renal pelvis. They are painful, especially during passage, and dangerous as a cause of chronic infection.
Calcium stones (oxalate, phosphate, others) are the commonest, and the majority of these patients have hypercalciuria (with or without hypercalcemia).
Hypercalcemia in a patient who first presents with kidney stones is probably due to a parathyroid adenoma.
Causes of hypercalciuria with normal serum calcium include milk-alkali syndrome (old ulcer cure; Am. J. Med. 99: 102, 1995), vitamin D abuse, mild sarcoidosis, dolomite pills (* polished rocks sold in health-food stores and promoted to athletic coaches "to prevent cramps due to hypocalcemia"), ketogenic anti-epileptic diets that include water restriction (J. Urol. 164: 464, 2000), and mild renal calcium or phosphate leaks. The recommended calcium supplementation for osteoporosis prophylaxis isn't enough to cause kidney stones in normal people.
Some calcium oxalate stone formers have increased absorption of oxalic acid from the gut (i.e., terminal ileitis, intestinal bypass, spinach gourmets); others are vitamin C abusers; still others have a minor inborn error of metabolism (* usually glycolic aciduria).
However, most calcium stone formers seem to have more calcium in the urine for no clear reason, i.e., probably they just absorb calcium from their gut too well. (This runs in families.) It's now generally accepted that most calcium stone-formers can be found to have one of the metabolic kinks if you care to look (NEJM 363: 954, 2010). You'll be treating these people with diuretics (if nothing else, diuretics force them to drink more water!) and magnesium citrate (citric acid solubilizes calcium.)
* Worth watching is NHERF1, the gene for protein controlling phosphate resorption; mutations here predispose to kidney stones and osteomalacia (NEJM 359: 1128 & 1171, 2008). A severe autosomal recessive disease ("hereditary hypophatemic rickets with hypercalciuria") is at the NPT2c locus.
* Also worth mentioning: "Randall's plaques" are bits of calcium in the tubular epithelium of the papilla. Nobody understands what causes these or whether they are related to kidney stones (Urol. Clin. N.A. 40: 1, 2013).
* In 2007-2008, unscrupulous food producers in China added melamine to animal food and powdered infant milk to boost the assayed protein content. Melanine is lost in the urine, and it's been suggested that this is the cause of the sand-like kidney stones seen in some of these children; because of selection bias and the composition of the stones being uncertain, the real risk remains unclear (NEJM 360: 1067 & 1139, 2009; Lancet 373: 353, 2009).
* Recreational use of ketamine causes kidney stones / hydronephrosis. The metabolites turn into a sludge in the tubules (Clin. Kid. J. 1: 310, 2008).
Magnesium ammonium phosphate stones ("* struvite stones", "triple stones", "infection stones") often result from urinary tract infection by urea splitters like proteus.
Most staghorn calculi, which fill the renal pelvis, are magnesium ammonium phosphate.
Only half of uric acid stones are associated with hyperuricemia. (* Ask them if they are taking any medications, especially non-steroidal anti-inflammatory agents.)
Cystine stones indicate cystinuria, which requires pharmacologic treatment.
* Triamterene stones (from patients on "Dyazide"): An intriguing subject. The stones are generally mixed. Also Acyclovir stones and Indinavir stones.
Future radiologists: calcium oxalate, calcium phosphate, and cystine stones are radio-opaque. Magnesium ammonium phosphate stones are slightly radio-opaque, and uric acid stones are radiolucent.
Future clinical pathologists: The lab does chemical analysis of calculi using many interesting procedures.
* Although "pain isn't real", you might enjoy reading about Mary Baker Eddy's experience with "the illusion of kidney stones", for which she took morphine. She was the founder of an anti-medical sect; the fact that she found so many followers simply reminds us that, in her era, mainstream medicine (without antibiotics, without safe surgery, without high-therapeutic-index medications, etc.) had much less to offer than it does nowadays.
* This is a good place to mention the uncommon and mysterious "loin pain hematuria syndrome". Patients have gross or at least microscopic hematuria, intractable loin pain, and seem to benefit from having the kidney moved surgically (autotransplantation -- could this work simply by severing the pain fibers to the kidney?) See J. Urol. 160: 1232, 1998.
KIDNEY TUMORS: Heidelberg classification J. Path. 183: 131, 1997.
|
|
THE BENIGN TUMORS
Little yellow nodules composed of clear cells are fairly common incidental findings at autopsy. They resemble miniature, low-grade renal cell cacinomas. Their natural history is unknown. If it were met in real life, each would be treated as a clear-cell renal cell carcinoma.
Papillary adenomas ("true adenomas") resemble small (less than 5 mm) papillary RCC's histologically and cytogenetically.
Hemangiomas (really hamartomas, not tumors) can bleed on-and-off.
* Laser cure: South. Med. J. 88: 759, 1995.
* Renal medullary fibromas are common incidentalomas at autopsy.
ANGIOMYOLIPOMAS, hamartomas of smooth muscle, vessels and fat, can occur in anyone, but are very common in tuberous sclerosis and its variant sporadic lymphangioleiomyomatosis. They may be troublesome by their size.
* Sirolimus, the mTOR signal inhibitor, for angiomyolipomas: NEJM 358: 140, 2008; Arch. Dis. Child. 95: 391, 2010. Everolimus: Lancet 381: 817, 2013.
{25360} angiomyolipoma, gross; aorta is small
because this is from a child
{25361} angiomyolipoma
![]() Renal angiomyolipoma
Renal angiomyolipoma
H&E
Wikimedia Commons
* Hemangiopericytomas (JGA-oma, renin-oma) are rare, make renin (rhomboid granules), cause high blood pressure (J. Urol. 146: 1607, 1994; reviews J. Urol. 153: 1781, 1995; Am. J. Clin. Path. 116: 854, 2001). Don't worry about the morphology.
Hürthle cell adenomas ("oncocytomas") turn up commonly as incidental findings on scans, and are often multiple. Their behavior is almost always benign unless there's obvious cellular atypia (* I'd rather call these oncocytic RCC's). They have a characteristic pattern of chromosome breaks, always have a central stellate scar, etc. See Am. J. Surg. Path. 21: 871, 1997. This was the first neoplasm to be linked to an abnormality of mitochondrial (rather than nuclear) DNA: Am. J. Path. 134: 967, 1989. The cell of origin is distal tubule (* some say "intercalated cells"; future pathologists: stain with Galectin-3: Arch. Path. Lab. Med. 134: 90, 2010). Some say it's best to take them out if you find them, as they continue to grow (J. Urol. 186: 1218, 2011).
* Let us worry about "mesoblastic nephroma", a baby's tumor that mimics a leiomyoma and raises renin and calcium -- excellent prognosis.
* "Metanephric adenoma" (along with its variant "metanephric adenofibroma") is a benign tumor adults that looks like a Wilms' tumor but without the anaplasia. The cells are small with hyperchromatic nuclei and have very little cytoplasm It's fine to leave it alone unless it produces erythropoietin and causes polycythemia.
CLEAR-CELL RENAL CELL CARCINOMA ("hypernephroma," "adenocarcinoma", "Grawitz tumor", "RCC"; Lancet 373: 1119, 2009; all renal cancers Arch. Path. Lab. Med. 133: 1026, 2009 and Arch. Path. Lab. Med. 135: 92, 2011 -- the latter for immunohistochemistry folks)
{21021} renal cell carcinoma, gross
{21022} renal cell carcinoma invading the vein
{09754} renal cell carcinoma, gross
{09788} renal cell carcinoma, gross
{09823} renal cell carcinoma, gross
{17204} renal cell carcinoma, microscopic;
the nuclei look deceptively benign
{17207} renal cell carcinoma, microscopic
{35582} renal cell carcinoma, microscopic
{00080} renal cell carcinoma, clear cells
|
|
|
|
|
|
|
|
![]() Renal clear cell carcinoma
Renal clear cell carcinoma
Low-grade
Wikimedia Commons
Unless otherwise specified, "renal cell carcinoma" refers to the clear-cell type.
Clear-cell RCC is a common, capricious cancer infamous for the many ways in which it can present clinically.
* Whether it's wise to call it "clear cell RCC" has been debated, since other kidney cancers, notably a distinctive rarity that's always papillary and is genetically unrelated to classic RCC, can be composed primarily of clear cells. But the term "conventional RCC" didn't catch on.
The classic triad is fever, hematuria, and flank pain. You actually see this occasionally in these patients, but don't expect a renal cell carcinoma to "make a typical presentation".
Or the tumor may present as hypercalcemia (J. Urol. 145: 248, 1991), amyloidosis, polycythemia, high blood pressure, estrogens, Cushing's, eosinophilia, leukemoid reaction, or as metastases (especially as a single metastasis in the head and neck). A few make erythropoietin and cause polycythemia.
Clear-cell renal cell carcinomas are usually bulky, yellow cancers that occur in middle-aged or elderly patients.
Tobacco is a known risk factor (update Cancer 118: 1795, 2012). Obesity is a minor risk factor in both genders (NEJM 343: 1305, 2000).
* Aspirin, touted as a risk factor, didn't pan out; others seem even softer Cancer 75: 2552, 1995.
Trans-stygian kidneys often give rise to renal cell carcinomas.
* The 1997 TNM staging system (Cancer 91: 354, 2001) defined tumors up to 7 cm -- which would be huge if they occurred elsewhere -- as at the lowest stage with a good prognosis if there are not positive lymph nodes or metastases (about 90% chance at a cure). If it's 4 cm or less, they do even better (J. Urol. 165: 1103, 2001). The newest TNM system validated: J. Urol. 173: 1889, 2005.
In both familial and sporadic cases, the anti-oncogene (VHL) at the Von Hippel Lindau locus is almost always deleted (Am. J. Hum. Genet. Dec. 1994; JAMA 273: 564, 1995).
The cell of origin is the proximal tubular cell, and there are always clear cells.
The clear cells are, rich in both lipid and glycogen, with no vacuoles; they may look wildly anaplastic or deceptively benign.
Clear-cell RCC's stain famously for vimentin, and are usually positive for epithelial membrane antigen and low-MW cytokeratin. Conveniently, there's a marker for proximal tubular differentiation (i.e., clear-cells and papillaries) that is almost always positive in a true-clear cell RCC, called "RCC / RCCMa"; these will also light up with CD10. Today we are using carbonic anhydrase IX (CA-IX), an enzyme downstream from the VHL product, consistently lights up renal cell carcinomas (clear cell RCC's usually light up most intensely); most other tumors do not show it. Update Arch. Path. Lab. Med. 133: 1026, 2009.
* The new marker for kidney epithelial cells / tumors is PAX8 (J. Clin. Path. 65: 254, 2012).
The Fuhrman grading of system for RCC's is based entirely on nuclear morphology (Am. J. Surg. Path. 6: 655, 1982 for the classic article; J. Clin. Path. 47: 324, 1994; update J. Urol. 177: 430, 2007). Molecular markers prognosticate in metastatic disease (J. Urol. 173: 1496, 2005).
* A variety of other histologic variables have been found to correlate with prognosis, even the prevalence of pericytes on the vessels (Cancer 119: 313, 2013) and staining for EZH2 (makes it more likely to metastasize Arch. Path. Lab. Med. 137: 1326, 2013).
* An alternate grading system looks only at nucleoli and necrosis (Am. J. Surg. Path. 37: 311, 2013).
Since only about 10% of RCC patients will get a second one, you can probably safely treat the little (3 cm) ones with partial nephrectomy (J. Urol. 153: 620, 1995). In fact, older folks with smallish renal cell carcinomas actually live longer and better if you do a partial nephrectomy (JAMA 307: 1629, 2012).
* Future radiologists: An antibody that lights up renal cell carcinomas: Lancet Oncol. 8: 279, 2007.
The tumor typically metastasizes by the bloodstream (unusual for a carcinoma; the other three carcinomas that tend to do this are hepatocellular carcinoma, adrenal cortical carcinoma, and follicular carcinoma of thyroid).
About 50% of clear-cell RCC's invade and grow up renal vein, and can extend right through the tricuspid valve, causing right-sided heart failure. (Such tumors are often successfully cured by removal: Am. J. Surg. 168: 205, 1994; a majority seem to be cured surgically J. Urol. 155: 448, 1996.) This shouldn't surprise you if you remember basic cancer biology. If the tumor has grown this big without the patient dying of metastases, the tumor hasn't yet acquired the ability to metastasize.
* Massive embolization of tumor itself to the pulmonary arteries is also infamous (Ann. Thoracic Surg. 61: 708, 1996). However, it's not ominous, and can be removed for the patient's symptomatic relief and sometimes cure: J. Thorac. Cario. Surg. 139: 320, 2010.
* "Because kidney is mesoderm", it's not uncommon for sarcoma to develop within a renal cell carcinoma, and metastasize as such: Cancer 116: 574, 2010. The molecular biology of "sarcomatoid renal cell carcionoma" is being worked out: J. Clin. Path. 64: 1088, 2011.
Five year survival may be about 70% for surgically-excised localized tumors, but survival is poor if tumor invades perinephric fat or metastasizes.
Cancer chemotherapy was famously unsuccessful for clear-cell RCC, but today we're having some success with combination of bevacizumab (the anti-VEGF antibody) and interferon-alpha (Lancet 370: 2103, 2007). Adding the very-toxic but powerful interleukin 2: Lancet 375: 641, 2010. Sunitinib (an anti-VEGF molecule): Cancer 116: 5389 & 5400, 2010 has been joined by sorafenib and pazopanib. The mTOR (rapamycin pathway) inhibitors temsirolimus and everolimus are new players.
Unlike most cancers, renal cell carcinoma will occasionally remit spontaneously (J. Urol. 152: 156, 1994), and nobody has a clue how. Or the metastases may remit after the primary is removed (J. Urol. 153: 751, 1995).
Better news: It's fairly common to find small renal masses on scans. Many of these are probably renal cell carcionomas. For patients who are not good surgical candidates, radiofrequency ablation is often used. Nowadays, it's clear that the vast majority of these folks get cures (Cancer 116: 3135, 2010).
And now there is talk about "durable cmplete responses" (dare we say "cures"?) with the new targeted biological protocols that include interleukin 2. Stay tuned.
|
|
In 2004, the World Health Organization published a system for distinguishing about forty primary renal tumors of adults from common clear-cell renal-cell carcinoma. You will go crazy if you try to learn them at your level. Students with a special interest may see the list in Arch. Path. Lab. Med. 131: 1234 &/or 1290, 2007, but I'd encourage you to visit this only if you have a patient who presents a challenge.
Mostly these are distinguished using immunoperoxidase techniques when the pathologist notices "this doesn't look like a common clear-cell RCC." PAX2 and RCC-Ma sensitive-and-specific immunostains for something of kidney origin, regardless of subtype (except maybe chromophobe carcinoma) -- you'll use these on metastases from unknown origin.
PAPILLARY RENAL CELL CARCINOMAS make up about 10% of primary adult kidney cancers. They're common in the trans-stygian kidneys of dialysis patients.
If the cells are clear but it's papillary, the prognosis is more ominous: J. Urol. 185: 30, 2011.
A proposal to grade papillary carcinomas by their nucleoli didn't show predictive value as well as Furham grade (J. Urol. 183: 2143, 2010).
* Future pathologists only: Papillary renal cell cacinomas light up with alpha-methylacyl-CoA-racemase (AMACR) (Am. J. Surg. Path. 28: 69, 2004) and often show lipid-laden macrophages within their papillae.
The 1% of primary kidney cancers that are of COLLECTING DUCT ORIGIN (* Bellini cancers / "collecting duct carcinomas") tend to be mucin positive, desmoplastic, hypovascular, ulex positive, and present hobnail cells (Urology 47: 921, 1996; Hum. Path. 21: 449, 1990; Arch. Path. Lab. Med. 123: 638, 1999). You'll see carcinoma in situ in the nearby collecting ducts. Cytogenetically, there are multiple monosomies. The cancer is quite aggresive. Update J. Urol. 182: 2595, 2009.
TRANSLOCATION-ASSOCIATED RCC is a distinct entity with t(Xp11.2;17). Pathologists recognize it by the expression of TFE3 on the nuclei and it cannot be diagnosed without immunostain or cytogenetics. It resembles alveolar soft-part sarcoma, caused by a similar X-chromosome translocation. Update Am. J. Clin. Path. 137: 761, 2012.
RENAL MEDULLARY CARCINOMA, a vicious variant that grows and spreads fast and simply does not respond to chemotherapy, appears almost exclusively in folks with sickle cell trait or disease (why?): Radiology 195: 83, 1995, Cancer 78: 128, 1996; J. Urol. 157: 2246, 1997; Arch. Path. Lab. Med. 127: e135, 2003. The diagnosis is clinched by noting that the nuclei do not stain with INI1, which normally lights up nuclei of the common kidney cancers (Am. J. Surg. Path. 33: 542, 2009).
CHROMOPHOBE RENAL CELL CARCINOMA, a tan (not yellow) cancer with sheets of cells with raisin nuclei, clear perinuclear areas (odd mucopolysaccharide in vesicles plus mitochondria accumulate around the nuclei) and pink-staining rims, the largest cells being around the blood vessels, has a more favorable prognosis (J. Urol. 154:964, 1995). If they metastasize, it's usually to liver (Am. J. Clin. Path. 111: 539, 1999; Cancer 78: 1756, 1996). Vimentin is negative, there are several curious antigens that distingish it from clear-cell RCC, and Claudin-7 is now seen as fairly specific marker for the chromophobe: Arch. Path. Lab. Med. 131: 1541, 2007. There's a syndrome (* gene BHD1), and a eosinophilic / Hurthle-cell variant (Mod. Path. 18: 161, 2005). Prognosticating Am. J. Surg. Path. 36: 851, 2012. * Colloidal iron stains stronly between the cells -- nobody knows quite why.
"Mixed epithelial and stromal tumor" is the best name so far for a curious, generally benign tumor of women that has ovarian stroma and lights up for estrogen and progesterons receptors (Arch. Path. Lab. Med. 133: 1483, 2009).
A variant with large eosinophilic cells arises in trans-stygian kidneys (Mod. Path. 19: 780, 2006).
We are now distinguishing the adult kidney cancers from each other and from benign mimics with great accuracy, thanks to gene technology on tiny samples: J. Urol. 176: 1957, 2006.
WILMS TUMOR ("nephroblastoma"): One of the commonest pediatric solid tumors (peak age 1-4 years). Pathology update J. Clin. Path. 63: 102, 2010 -- suggests further refinement of the grading system (which is presently "Wilms" or "Anaplastic Wilms").
{25369} Wilms, histology
{25370} Wilms, histology
{25373} Wilms, some second-rate rhabdomyoblasts
A tumor of variable histopathology, often a carcinosarcoma, often with embryonic renal blastema, mesenchyme, muscle, sometimes cartilage, plus attempts to form tubules and glomeruli. Usually all three components are present (blastema, tubules, and mesenchyme/stroma-like/muscle.)
The histopathologic subclassification and prognostication of Wilms tumor has been carefully worked out -- leave it to us to worry about (Urol. Clin. N.A. 27: 435, 2000).
Patients often have aniridia, hemihypertrophy, and/or rests of undifferentiated blastema.
* In sub-Saharan Africa, where Wilms tumor is very common in some locales, an environmental factor is almost certainly operating, but so far it has escaped identification (Am. J. Trop. Med. 54: 343, 1996).
The ones with aniridia are carrying a deletion of 11p13, called the WT-1 (formerly the WAGR) locus. One portion confers aniridia, while Wilms' tumors themselves become homozygous for a nearby portion (i.e., the place where the anti-oncogene is located).
* Denys-Drash, a syndrome with only kidney problems and a Wilms tumor risk, is also inherited at this locus.
* Update on the molecular biology of Wilms tumor: Urol. Clin. N.A. 27: 423, 2000. One interesting finding is that the Wilms' tumors that make cartilage, muscle and stuff have less WT1 mRNA, demonstrating that WT1 is responsible for proper differentiation (no surprise, Am. J. Path. 140: 781 & 1031, 1992).
Even with metastases, chemotherapy often cures Wilms' tumors, but the prognosis of very aneuploid tumors is still guarded. Protocols will continue to change.
|
|
* OTHER PEDIATRIC KIDNEY TUMORS
"Clear cell sarcoma" of the kidney ("bone-metastasizing renal tumor of childhood") is another pediatric cancer, more ominous than Wilms, notable for a tendency to metastasize to bone / skull (unlike Wilms), and with about a 75% cure rate at best.
"Rhabdoid tumor" is a pediatric tumor (around age 18 months) made of cells that look like rhabdomyoblasts, but that do not stain for myoglobin. It starts in the medulla. The prognosis is usually unfavorable. There is a trademark 22q11 translocation. Arch. Path. Lab. Med. 127: 371, 2003; Arch. Path. Lab. Med. 131: 102, 2007.
"Congenital mesoblastic nephroma" is the common "kidney tumor of the newborn". It's generally well-behaved (Arch. Path. Lab. Med. 128: 929, 2004). There's an aggressive "cellular" variant that is actually a pediatric fibrosarcoma, sharing ETV6-NTRK3 gene fusion with other infantile fibrosarcomas (Am. J. Path. 153: 1451, 1998).
UROTHELIAL CARCINOMA ("transitional cell" carcinoma of renal pelvis; "upper urinary tract urothelial carcinoma" / UTUC)
Tumor of older adults; much less common than renal cell carcinoma. Usually arises multifocally along with cancers in urinary bladder.
Similar to bladder cancer; papillary growth is usual, prognosis is guarded especially if there's invasion. As in bladder, some tumors are much more aggressive than others.
* In the unlikely case that the pathologist cannot tell a urothelial cancer from a renal cell carcinoma, the former tends to stain with uroplakin III, p63, and thrombomodulin. Kidneys of renal parenchymal origin usually won't.
* If the patient is under age 60, think of Lynch's microsatellite instability syndrome.
{09778} urothelial carcinoma of renal pelvis
{18790} urothelial carcinoma of renal pelvis
{39762} urothelial carcinoma of renal pelvis
{39844} urothelial carcinoma of renal pelvis --
this is invading the cortex
|
|
![]() Urothelial carcinoma
Urothelial carcinoma
Renal pelvis
WebPath Photo
TRANSPLANT PATHOLOGY
ACUTE TUBULOINTERSTITIAL ("CELL-MEDIATED") REJECTION (i.e., host T-cells are attacking the kidney)
CHRONIC REJECTION (i.e., after 3 months or so, and increasing as the years go by)
World Kidney Day is the second Thursday in March. (Good site -- and a good cause.)
Your lecturer believes that most people want to help strangers when they can and when it costs them nothing. When dialysis was instituted in the 1960's, it was with the expectation that most people would want to donate their deceased loved-one's kidneys and that no one would have to be on dialysis for long. Sadly, a mix of ideologues and tort lawyers (and perhaps third-party payers) has made this much more difficult than it should be. We look with hope to countries like Spain and Belgium, where the doctor will take the decedent's kidneys unless there's a previous opt-out (Lancet 378: 1356, 2011).
* SLICE OF LIFE REVIEW
{03184} renal tubules composite of micro, normal vs. cell swelling
{03184} renal tubules composite of micro, normal vs. cell swelling
{11813} kidney, normal cut surface
{14919} kidney, cortex & medulla
{14920} kidney, cortex & medulla
{14921} kidney, cortex & medulla
{14922} kidney, cortex & medulla
{14923} kidney, cortex
{14924} kidney, cortex
{14925} kidney cortex, (glomerulus)
{14926} kidney cortex, (glomerulus)
{14927} kidney, tubules (distal & proximal)
{14928} kidney, tubules (distal & proximal)
{14929} kidney, medulla (collecting tubules)
{14930} kidney, medulla (collecting tubules)
{14931} kidney, medulla collecting duct
{14932} kidney, medulla collecting duct
{14933} juxtaglomerular apparatus
{14934} juxtaglomerular apparatus, kidney
{14935} glomerulus, (vascular pole)
{14936} glomerulus, (vascular pole)
{15316} kidney, cortex
{15317} kidney, medulla
{15318} kidney, papilla
{15319} kidney, medullary ray
{15320} kidney, medulla
{15321} kidney, macula densa and distal tubule
{15322} kidney, area cribrosa
{15323} kidney, medullary ray
{15324} kidney, cortex
{15572} kidney, normal
{15573} kidney, normal
{15574} kidney, normal
{15834} kidney, normal neonatal
{16657} kidney, normal
{16658} kidney, normal
{16723} kidney, normal
{16724} kidney, almost normal -- the cortex is a bit pale, suggesting terminal ATN
{16729} glomerulus, normal
{16730} glomerulus, normal
{16732} glomerulus structure, normal
{16733} glomerulus structure, normal
{16735} glomerulus capillary structure, normal
{16738} glomerulus capillary structure, normal
{16739} glomerulus capillary structure, normal
{16740} glomerulus ultrastructure, normal
{16741} glomerulus ultrastructure, normal
{16744} glomerulus ultrastructure, normal
{16748} glomerulus, normal
{16762} glomerulus ultrastructure, normal
{16763} glomerulus, normal
{16859} kidney, normal
{17027} glomerulus, normal
{17028} glomerulus, normal
{17149} kidney, normal-1303
{17150} kidney, normal-1303
{17534} kidney, normal
{17576} kidney, normal
{17577} kidney, normal
{19798} kidney, normal
{19799} kidney, normal
{19800} kidney, normal
{19801} kidney, normal
{19802} kidney, normal
{19803} kidney, normal
{20924} kidney, papilla
{20925} kidney, medullary ray
{20926} kidney, collecting duct
{20927} kidney, distal tubule
{20928} thick loop of henle, kidney
{20929} kidney, collecting duct
{20932} kidney, macula densa
{20933} kidney, parietal cells
{20934} kidney, proximal tubule
{20935} kidney, distal tubule
{20936} kidney, collecting duct
{20937} kidney, proximal tubule
{20938} kidney, distal tubule
{29587} kidney, normal biopsy
{33307} glomerular disease, normal?
{34216} glomerulus, normal
{34282} glomerulus, normal
{34291} glomerulus, normal
{34306} glomerulus, normal
{39473} glomerulus, normal
{46462} cilia in collecting tubule of kidney, normal
{46463} glomerulus, normal
{46526} proximal tubules, normal
{46527} glomerular capillary, normal
* I SAW A MAN PURSUING
I saw a man pursuing the horizon;
Round and round they sped.
I was disturbed at this;
I accosted the man.
"It is futile," I said, "You can never -- "
"You lie," he cried, And ran on.
-- Stephen Crane
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about kidney pathology to consult these standard textbooks.
In my notes, the most helpful current journal references are embedded in the text. Students using these during lecture strongly prefer this. And because the site is constantly being updated, numbered endnotes would be unmanageable. What's available online, and for whom, is always changing. Most public libraries will be happy to help you get an article that you need. Good luck on your own searches, and again, if there is any way in which I can help you, please contact me at scalpel_blade@yahoo.com. No texting or chat messages, please. Ordinary e-mails are welcome. Health and friendship!

| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |


 Kidney and Lower Urinary
Kidney and Lower Urinary
 Immunotactoid glomerulonephritis
Immunotactoid glomerulonephritis
