Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
verify here. |
Cyberfriends: The help you're looking for is probably here.
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site at
www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()
This chapter is absolutely pivotal to your learning medicine. The handout, which is relatively short, is really all mastery material. You need to learn the content at the recall level.
Recognize what things that happen to a person result in inflammation, and distinguish its acute and chronic phases.
Give a full account of the stereotyped processes of acute inflammation. Explain the pathophysiology underlying the classic "rubor, calor, dolor, and tumor", and a complete account of vascular caliber and permeability changes during acute inflammation.
Give a complete account of white cell behavior in acute and chronic inflammation. Tell what white cells are (and aren't) recruited in various kinds of inflammation, and in response to which invaders. Tell the means by which white cells are recruited, the weaponry they carry, and how they find and destroy invaders.
Explain when and how pus forms, and account for its familiar properties and variable appearances. Mention factors that interfere with leukocyte function.
Describe the acute phase reaction and the physiology of the erythrocyte sedimentation rate. Give a short account of the "systemic inflammatory response" and mention why it's deadly.
Describe the role of mononuclear phagocytes in inflammation. Tell how and when granulomas form, and why they are important.
Give a good basic account of how tissues regenerate, injuries heal, and scars form. Given the name of a cell, tell whether it is labile, stable, or permanent, and why it matters. Explain how fibrosis forms in chronic inflammation. Describe how a fibrin meshwork is transformed into a fibrous scar. Distinguish healing by primary and secondary intention, and cite the factors that promote and oppose good wound healing.
Describe the activities of the following mediators of inflammation and/or healing, and when an activity is mentioned, remember which molecule or molecules mediates the effect:
bradykinin
C3a
C3b
C5a
histamine
IgE
interferon
interleukin 1
leukotrienes
membrane attack complex
platelet-derived growth factor
prostacyclin
prostaglandin E2
serotonin
transforming growth factor β
thromboxane A2
Use each the following terms properly, and recall the term, given the definition:
abscess
acute inflammation
acute phase reaction
adhesion molecules
arachidonic acid
chemokinesis
chemotactic agent
chemotaxis
chronic inflammation
contact inhibition
degranulation
empyema
emigration
eosinophil
erosion
exuberant granulation
exudate
fibrin/fibrinous
fibrinogen
fibrous/fibrosis
free radicals
granulation tissue
granuloma
hyperemia
infection
inflammation
keloid
labile cell population
left shift
leukocyte
leukemoid reaction
leukocytosis
lymphocyte
lysosomes
macrophage (AKA...)
margination
myeloperoxidase
neutrophil
neutrophilia
opsonization
organization
permanent cell pop.
phagocytosis
plasma cell
pseudomembrane
purulent
pus
regeneration
resolution
scar
stable cell population
suppurate
transudate
ulcer
Describe how problems with inflammation cause or exacerbate the following illnesses:
Chediak-Higashi syndrome
chronic granulomatous disease
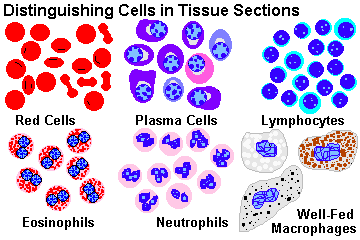 Be sure you can recognize each of the following under the microscope:
Be sure you can recognize each of the following under the microscope:
edema (when obvious)
eosinophils
fibrin
giant cells (both types)
granulation tissue
granulomas
neutrophils
macrophages
mast cells (when stained)
plasma cells
pus
red cells
scar tissue
ulcers
Use and apply the common suffixes for surgical operations properly.
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- Inflammation
Lectures follow Textbook
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- Repair
Lectures follow Textbook
QUIZBANK
Inflammation (all)
Healing (all)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LEARN FIRST
ACUTE INFLAMMATION is a stereotyped response to recent or ongoing injury. Although the process is complex, the principal features are dilation and leaking of vessels, and involvement of circulating neutrophils.
You can recognize neutrophils in tissue sections by their segmented nuclei.
PUS is neutrophils plus
liquefaction necrosis![]() .
Usually, the neutrophils themselves
caused most of the necrosis.
.
Usually, the neutrophils themselves
caused most of the necrosis.
CHRONIC INFLAMMATION ("late-phase inflammation") is a response to prolonged problems, orchestrated by T-helper lymphocytes. It may feature recruitment and activation of T- and B-lymphocytes, macrophages, eosinophils, and/or fibroblasts. Again, the process is complex. You will recognize lymphocytes in tissue section by their small, "blue button" nuclei.
GRANULOMAS are seen in certain chronic inflammation situations. They are clusters of macrophages that have stuck tightly together, typically to wall something off. Such macrophages are called epithelioid cells. You will recognize granulomas in tissue sections by their characteristic appearance, or the presence of giant cells.
FIBRIN is fibrinogen released from damaged vessels, and activated by the clotting cascades when blood meets tissue juices. Fibrin forms the meshwork that controls bleeding, and then becomes the framework for fibroblasts and angioblasts that will form the SCAR. Until the new scar is complete, the whole meshwork of immature scar is called GRANULATION TISSUE. When the scar has matured, it contracts.

|
What do scars do? They heal. No, wait. That's wounds. Scars..."
Scars contract. The Panda's right, too -- they can fade. |
|
INTRODUCTION
WAR IS THE METAPHOR FOR INFLAMMATION. Both are unpleasant facts of life. Both are more-or-less stereotyped responses to outside threats. There are specialized troops (white cells), including high-risk-commandos (neutrophils), long-term siege armies (granulomas), and many others. There are supply routes (vessels), communications and intelligence (mediators), and a huge array of lethal weapons (inflammatory enzymes). In war as in inflammation, there will be damage to both the enemy and to friendly forces, and there will very likely be severe damage to the battlefield itself. Despite idealistic rhetoric about "the laws of war", when the fighting starts, there is really only one law for the soldiers: "Kill your enemy." Like it or not, if you want peace, you must be prepared to fight under certain conditions. Like it or not, if you want to be healthy, your body must be able to mount an inflammatory response. Force will always rule our world. Our best hope is that this will be the force of good laws. And the best for which we can hope from the inflammatory response is that, for most of our lives, it will do us more good than harm. |

|
"Big Robbins" has defined INFLAMMATION as "the reaction of vascularized living tissue to local injury". Inflammation is the name given to the more-or-less stereotyped ways our tissues respond to noxious stimuli, with blood vessels and white blood cells as its twin centerpieces and a host of proteins as actors. Inflammation "destroys, dilutes, or walls off the injurious agent" and sets in motion the limited powers of the body to heal itself. Inflammation and repair can and do themselves damage the body.
Strictly speaking, "immunity" is all the things the body does in defense against invaders (growing skin, making neutrophils, etc., etc.) As used today, the unmodified word IMMUNITY refers to the activities coordinated by B ("humoral immunity") and T ("cellular immunity") lymphocytes and monocytes/macrophages.
Beginning medical students have a tendency to equate "inflammation" and
INFECTION, at least
unconsciously. Now is the time to understand why this is a mistake. Several infectious diseases feature no inflammation
(Creutzfeldt-Jacob disease, yellow fever, and many of the opportunistic infections in AIDS are only
three examples.) Noxious, non-infectious things that produce inflammation include
TRAUMA, RADIATION injury, various POISONS, chemical or thermal BURNS,
TISSUE NECROSIS itself (except
apoptosis -- the molecular mediators are unknown but are probably
nucleic acid breakdown products: J. Clin. Inv. 120: 1939, 2010),
and any of the four major types of IMMUNOLOGIC INJURY. (You'll learn about all of these soon
enough.) A sunburn and a red scratch are inflammation, just like mosquito bites, pimples,
plague![]() and
leprosy
and
leprosy![]() .
.
Obviously, there are differences among inflammatory reactions. ACUTE INFLAMMATION is almost completely stereotyped -- over minutes to a few days, blood vessels widen and disgorge protein, and neutrophils leave the bloodstream and rampage through surrounding tissues. CHRONIC INFLAMMATION is more variable, with variable participation by lymphocytes, plasma cells, macrophages, and healing cells (fibroblasts and angioblasts).
WHOLE BODY INFLAMMATION, formerly a popular term used especially by surgeons for the patients who they could not save, is going out of fashion in favor of MULTIPLE ORGAN FAILURE or SYSTEMIC INFLAMMATORY RESPONSE SYNDROME ("SIRS") or UNCONTROLLED IMMUNE RESPONSE (Am. Heart. J. 156: 1065, 2008) or STRESSED-HOST SYSTEMIC INFLAMMATION (Surg. Clin. N.A. 89: 311, 2009).
When it is caused by bacterial infection of the bloodstream, it's called SEVERE SEPSIS (JAMA 303: 2495, 2010; the current use seems to be that "sepsis" still means bacteria actually multiplying in the bloodstream).
* Defining "SIRS" -- i.e., the point in sepsis at which the risk of death becomes substantially greater -- remains a statistician's challenge. It turns out that the more criteria are met, the greater the risk of death, without any sudden jump (NEJM 372: 1629, 2015). This challenges the concept.
* Especially in the elderly, we are now noticing that people suffer brain damage after sepsis out of proportion to what can be explained by poor brain perfusion. It is likely that the activation of the macrophages in the brain ("microglia") is a factor (suggested prophylaxis Lancet 375: 773, 2011).
Whatever you call it, in super-sick people, various cytokines increase tremendously in the bloodstream; this situation interacts with ischemia, free radical production, and leakage of heatshock proteins from cells to create a vicious downward cycle into irreversible shock. Venous return to the heart (i.e., venous responsivity) is compromised, perhaps myocardial function is depressed, etc., etc., etc., etc., etc. See Surg. Clin. N.A. 80: 885, 2000; Crit. Care Med. 28: 537, 2000; Crit. Care Med. 29(7S): S-99, 2001.
As pathologists, we recognize "multiple organ failure" clinically by a striking drop in the absolute lymphocyte count caused by apoptosis of these cells; lymphoid depletion is seen at autopsy (J. Imm. 174: 3765, 2005).
We'll mention the cytokines that are thought to be involved below.
"Subacute inflammation" does not describe a distinct pattern.
We suggest that you first try to understand inflammation at the light microscopic level. You are already acquainted with fibrinogen and fibrin from college biology. Only when you have a clear picture of acute inflammation, chronic inflammation, and wound repair should you go back and learn the host of molecular mediators that derive from cells and plasma.
The suffix that indicates inflammation is "-itis" (the plural is "-itides". Philologists: "-osis" means "full of".)
* The public recognizes inflammation, and the words "inflame" and "inflammatory" have found their way into journalism and law.
Terms for abnormal accumulations of fluid: A TRANSUDATE is protein-poor salt water squeezed through blood vessels by hydrostatic pressure, i.e., it has very little protein, and has the specific gravity of extracellular fluid, 1.010 or thereabouts. An EXUDATE is an abnormal, protein-rich fluid that has leaked out of inflamed vessels. It's easy to measure the higher protein content and higher specific gravity.
A body fluid (either an exudate or an area of
liquefaction necrosis![]() ) containing neutrophilic
leukocytes and necrotic debris is PUS. The preferred adjective to describe things with lots of pus is
PURULENT. To produce pus is to SUPPURATE. Pus that literally fills an important body cavity is
called an EMPYEMA. (This is most common in the pleural cavities.) If you've got a lot of pus, you
need it drained surgically.
) containing neutrophilic
leukocytes and necrotic debris is PUS. The preferred adjective to describe things with lots of pus is
PURULENT. To produce pus is to SUPPURATE. Pus that literally fills an important body cavity is
called an EMPYEMA. (This is most common in the pleural cavities.) If you've got a lot of pus, you
need it drained surgically.
Pus requires no description, but it is worth mentioning that pus is not always the same color or thickness.
Classic yellow pus
(as in a staphylococcal![]() boil) also includes some lipid from necrotic tissue,
hence the stronger yellow color.
boil) also includes some lipid from necrotic tissue,
hence the stronger yellow color.
Without necrosis (as in a
streptococcal![]() phlegmon), pus is more yellow-gray.
phlegmon), pus is more yellow-gray.
Pseudomonas bacteria make a pigment that imparts a blue-green fluorescent sheen to pus.
Clostridia produce extensive hydrolysis of tissue, and the discharge from a clostridial wound contains free lipid and other small molecules that impart a "dirty dish-water" look.
{26419} normal neutrophil in a smear; finely granular
cytoplasm and segmented, dark nucleus
{14704} normal neutrophil in a smear; finely
granular cytoplasm and segmented, dark nucleus
{10067} pus in an abscess, section; notice how neutrophils look different in sections and smears
{08979} histopathology of an acne pimple! Find two cross-sections of the keratin plug.
![]() Neglected peritonitis with
Neglected peritonitis with
neutrophils and necrosis
One of my autopsies
Increased interstitial fluid is called EDEMA.
HISTORICAL HIGHLIGHTS: "Big Robbins" lists, or might have listed....
Cornelius Celsus (ancient Rome) described RUBOR (redness), CALOR (heat -- this applies only to the skin), DOLOR (pain), and "TUMOR" (in those days it simply meant "swelling"; regrettably, teaching this useless Latin term generates much confusion among medical students today) as the "cardinal signs of inflammation".
* John Hunter (1793, the great early surgeon) first characterized inflammation as a nonspecific body response.
Rudolf Virchow added FUNCTIO LAESA (loss of function) as the fifth cardinal sign of inflammation, and his student, Julius Cohnheim, provided the basic studies of the pathologic microanatomy of inflammation.
* Elie Metchnikoff (1892) became the first to observe and study phagocytosis. (This is the same Metchnikoff who popularized yogurt as a "health and longevity food". He died at age 70.)
Paul Ehrlich developed the idea of humoral immunity early in the 20th century. (This is the same Ehrlich who developed the "magic bullet" for syphilis, and most of the stains we still use.)
Thomas Lewis demonstrated that inflammation is brought about by chemical mediators, most of which act locally.
ACUTE INFLAMMATION: A stereotyped response to most kinds of noxious stimuli. Something a part of the body does when it knows it's been hurt.
* Mega-review: Med. Clin. N.A. 81: 1, 1997 (still good); J. Clin. Inv. 118: 413, 2008 (inflammation as "a calculated response")
Textbooks describe "acute inflammation" as lasting from moments to a maximum of 1-2 days. This is a simplification, as anyone with a persistent pimple knows. (* Your handout author has lots of experience with this.) THE HALLMARKS OF ACUTE INFLAMMATION ARE (1) VASODILATION AND INCREASED VASCULAR PERMEABILITY; AND (2) ENTRY OF NEUTROPHILS INTO THE TISSUE.
The first event is TRANSIENT ARTERIOLAR CONSTRICTION, lasting a few seconds (if at all; scratch yourself and see) up to a few minutes (after a trivial burn -- you have probably noticed it takes a while for a minor burn to turn red.) This vasoconstriction helps control blood loss in case vessels have been severed.
![]() Neutrophils on
Neutrophils on
pap smear
Dave Barber MD, KCUMB
|
|
When the arteriolar constriction phase is over, THE ARTERIOLES DILATE AND STAY DILATED as long as acute inflammation continues. This produces the redness and (since heart's blood is warmer than exposed body parts) the sensation of heat. The slightly increased pressure that this causes in the capillaries may produce some transudation of fluid into the tissue spaces, but this cannot be a huge effect (if it were, blushing would cause impressive edema).
HYPEREMIA is a generic term for extra blood in an organ due to dilation of the arterioles. More about this soon.
Soon after injury, THE SMALL VESSELS (supposedly mostly the venules 20-60 microns) BECOME PERMEABLE TO SOME OR ALL OF THE PLASMA PROTEINS. This increases the osmotic ("oncotic") pressure of the interstitial fluid, water is drawn out of the vessels, and INFLAMMATORY EDEMA ("swelling") results.
As protein leaks out into the interstitial spaces, the local concentration of cells in the blood increases. Red cells pack small vessels ("red cell stasis"), neutrophils stick to endothelium, and the viscous blood flows more slowly ("stasis"). The water that follows the protein out of the vessels contributes to edema. Much of this fluid will return to the circulation only via the lymphatics.
The physicochemical changes that cause the increased permeability to protein are only partly understood. The key seems to be opening gaps in the intercellular junctions ("endothelial cell contraction"). Another factor seems to be loss of various polyanions from the basement membrane surrounding the endothelial cells. Of course, if the vessels are damaged by the first injury, or by the neutrophils, or are themselves regenerating, they will leak.
The worse the injury, the larger the protein molecules that can pass through the vessel walls. In the worst injuries (and, of course, if the vessel is severed), FIBRINOGEN escapes into the tissue fluids, and under these circumstances is certain to be transformed to FIBRIN (by your clotting cascade, of course).
Of course, the fibrin controls bleeding and provides a mechanical barrier. If needed, it will also serve as the framework while the new scar tissue will be laid down. When you are starting out, it is easy to confuse fibrin and collagen. "The difference between fibrin and collagen is the difference between a scab and a scar."
NOTE: When unqualified, the word FIBROUS means "composed of type I collagen". FIBRINOUS always means "composed of fibrin".
{10901} gonorrheal salpingitis; note tremendous swelling and redness of both oviducts
{46310} acute inflammation from a bacterial infection of the kidney
{18719} fibrinous ("bread and butter", better "ketchup on bread") pericarditis
Experimentalists use colloidal carbon (i.e., fine-grain India ink) to demonstrate the increased vascular permeability. In the classic experiments, we distinguished three separate phases of vessel leakage:
(1) The familiar IMMEDIATE-TRANSIENT RESPONSE begins at once, peaks at 5-10 minutes, and is over by 30 minutes. It involves only the venules, involves CONTRACTION AND SEPARATION OF ENDOTHELIAL CELLS, and is attributed to PROSTAGLANDINS, HISTAMINE, serotonin and a host of other chemical mediators.
(2) The more persistent, equally-familiar IMMEDIATE-SUSTAINED immediate-sustained reaction ("immediate prolonged") is seen only when the injury is severe enough to cause DIRECT ENDOTHELIAL CELL DAMAGE, and persists until thrombosis or regeneration ends it. Obviously this can affect any blood vessel.
(3) The DELAYED-PROLONGED LEAKAGE phenomenon is seen only after hours or days. Venules and capillaries exude protein, again because their junctions separate. (Other vessels' walls are too thick to exude much protein.) The prototype Is the swelling that accompanies a sunburn, radiation therapy, and all but the worst thermal burns. Long-mysterious, we now know that the underlying mechanism is APOPTOSIS OF THE ENDOTHELIAL CELLS.
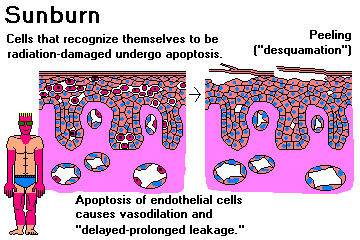
(4) White cells do some damage "just passing through".
(5) And of course new blood vessels (young scar, or cancers) are leaky.
The final key event in acute inflammation is the ACCUMULATION OF NEUTROPHILS ("polys", "segs"; nobody calls them * "microphages" nowadays) in the injured tissue. (Most of the time, these predominate for the first 24-48 hours after injury, and are more or less replaced by macrophages after this time.)
The laws of physics cause neutrophils to MARGINATE ("pavement", i.e., lie along the inner walls of vessels) whenever blood flow is slowed. They roll along for a while. ADHESION to the walls of vessels, especially venules, results when LEUKOCYTE ADHESION MOLECULES on the surface of the neutrophils interact with ENDOTHELIAL ADHESION MOLECULES on the endothelial cells.
Leukocyte adhesion molecules go by names such as LFA-1 and MO-1. These are members of a homologous set.
* Leukocyte adhesion deficiencies.... LAD I: Lack a CD11b/CD18 integrin; LAD II: lack the selectin-binding Lewis X glycoprotein. Newer variants Blood 97: 767, 2001. A dog model has been successfully treated using a viral gene therapy vector: Nat. Med. 14: 93, 2008.
* Right now, you'll go crazy trying to understand the role of the endothelial cell in various inflammatory diseases (update J. Immuno. 178: 6017, 2007). For now, simply understand that they're likely to be involved in several "idiopathic" diseases despite the fact that they seldom look like they're doing anything.
* For an easy-to-read update on the physiology of leukocyte recruitment, see J. Immuno. 180: 6439, 2008. For an account of the neutrophil and the vessel wall that will make you think of pinball (and predicts a new line of Rx's), enjoy Am. J. Path. 172: 1, 2008.
* We are just now starting to look at the interactions of neutrophils with red cells and platelets, especially while they're crawling along endothelial surfaces. For example, in an animal model of sickle-cell vascular disease, the neutrophils sandwich red cells, while in an animal model of transfusion-related lung disease, the neutrophils cling to platelets (both Nat. Med. 15: 384, 2009).
Endothelial adhesion molecules include ELAM-1 (for neutrophils) and ICAM-1 (for neutrophils, lymphs, and monos). Various mediator proteins increase the numbers of some or all of these.
* Stay tuned: Pain itself (i.e., stimulation of pain fibers) causes the local neutrophils to lose their ability to express L-selectin adhesion molecules. This seems to be one mechanism of feedback that limits the acute inflammatory response (Nat. Med. 5: 1057, 1999).
EMIGRATION ("diapedesis") of neutrophils from the vessels into the tissues occurs when the cells squeeze through the widened endothelial cell gaps, then get through the basement membrane by digesting it with enzymes. (Of course this damages the blood vessels, but the endothelial cells repair the damage soon enough.) The other white cells also leave vessels by this route.
Various chemical mediators cause CHEMOKINESIS (increased random locomotion) and CHEMOTAXIS (directional migration) of neutrophils and other cells. CHEMOTACTIC AGENTS include a plethora of bacterial breakdown products, complement components (remember C5a), and leukotriene B4. Most small molecules that are chemotactic for neutrophils are also chemotactic for macrophages and vice versa.
Chemotactic factors act on the cell membrane, signalling a poorly-understood process involving the cytoskeleton (remember CALMODULIN, the calcium-binding protein that polymerizes myosin, as a key player here) that eventually results in cell movement.
Certain lymphokines (factors produced by lymphocytes) and monokines (factors produced by monocytes / macrophages) are chemotactic for neutrophils and/or other white cells. Mast cells, activated in parasitic infestations and classic IgE-mediated allergy, release "eosinophilic chemotactic factor of anaphylaxis".
* Someone may ask you about key enzymes in the production of prostaglandins and leukotrienes. "Phospholipase A" releases arachidonic acid from a host of biologic membranes and is inhibited by glucocorticoids. "Cyclo-oxygenase" turns arachidonic acid into prostaglandins and is inhibited by aspirin. "5-lipo-oxygenase" turns arachidonic acid into leukotrienes.
Once they have found their way to the tissues, the neutrophils PHAGOCYTIZE things that shouldn't be there. They also DEGRANULATE, releasing enzymes into the interstitial fluid.
PHAGOCYTOSIS requires that the particle be recognized and attach to the neutrophil. Most particles must be coated (OPSONIZED) by IgG (subtypes 1 or 3) or C3b. There are receptors for both on the neutrophil surface. The particle will then be engulfed and a lysosome membrane fused with the phagosome membrane, causing digestion within the phagolysosome. (If only C3b is present in the opsonin, additional molecules will be required to trigger engulfment.) Some of the lysosomal enzymes will leak out of the neutrophil and into the intercellular fluid.
KILLING of phagocytized bacteria is mediated through the H2O2-myeloperoxidase-halide system and other, less-effective oxygen-dependent and oxygen-independent systems. Exactly how this happens is still under investigation (see for example a claim based on generating charge differentials across membranes in Ann. Rev. Immuno 23: 197, 2005). We retain ancient microbe-killing proteins including lysozyme and lactoferrin.
NEUTROPHIL PRODUCTS, including LYSOSOMAL ENZYMES, H2O2, free radicals, and ARACHIDONIC ACID METABOLITES are released during the process by "regurgitation during feeding", "frustrated phagocytosis" (i.e., the neutrophil tries to eat something too big, such as a huge immune complex or a splinter; it can't engulf it so it drools), and "cytotoxic release". The latter is a euphemism for stuff leaking out of dead cells.
You are already aware that neutrophils are programmed to undergo apoptosis in 72 hours. How this happens is now becoming clear (J. Imm. 175: 1232, 2005); it is now known to be delayed (though not abolished) in recruited neutrophils.
Notable exceptions to the "in the tissues it is neutrophils first, monocytes later" rule:
(2) In classic allergy, asthma, and in most worm infections, eosinophils dominate from start to finish;
(3) In typhoid fever, the predominant cells are always the macrophages;
(4) In most forms of acute dermatitis, lymphocytes are most abundant;
(5) In clostridial gas gangrene, don't expect to see any white cells;
(6) In many kinds of bacterial
infections (including some chlamydial![]() ones), there are few or no other cells besides the neutrophils.
ones), there are few or no other cells besides the neutrophils.
* In some chronic inflammatory situations, the number of mast cells increases and they can be more easily triggered; to what extent this contributes to disease is still mysterious (J. Allerg. Clin. Immuno. 127: 905, 2011).
Here's something to help you appreciate neutrophils and what they do to normal tissue. You know that after you've had a cold and runny nose for a few days, the skin at the inner edge of your nostril becomes cracked and sore. This is the effect of enzymes from the neutrophils that have responded to the virus having caused a bit of tissue injury. By contrast, in a runny nose from hay fever, there is no cell injury, hence there are no neutrophils, hence no injured skin on your nose.
Neutrophil defects worth learning now:
1. Too few circulating neutrophils ("neutropenia"; "agranulocytosis"), as in radiation injury, cancer therapy, drug sensitivity
2. Neutrophil adherence molecule defects, due to heredity, glucocorticoid administration, diabetes, or ethanol in the bloodstream.
3. Failure of neutrophils to move properly (notably in diabetes) or to respond to chemotactic stimuli
4. Failure of neutrophils to phagocytize (diabetics, people with complement or immunoglobulin deficiencies)
5. Defective microbial killing. This may be due to
6. Mixed defects. Remember that diabetes and glucocorticoids interfere with most white blood cell functions. In CHEDIAK-HIGASHI SYNDROME (a problem with membrane synthesis), there are too few neutrophils, they do not respond properly to chemotactic stimuli, and their (abnormally large) lysosomes fail to fuse with phagosomes.
* Nuclear medicine techniques to discover abscesses by following tagged neutrophils have largely given way to other imaging procedures.
Unless the injury is trivial, mediators produced by other cells will cause increased production and early release of neutrophils from the bone marrow. Increased neutrophils in the blood stream is NEUTROPHILIA (or, sloppy, "leukocytosis"), and the presence of young neutrophils ("bands", etc.) is called a LEFT SHIFT, after the column positions on the old hematologists' counting pad. You'll learn later how to tell real leukemia from a severe inflammatory response (LEUKEMOID REACTION).
{26191} mature neutrophils
{13655} mature neutrophils (lots of segments;
can you see the granules?)
{16186} electron micrograph of neutrophil showing granules
{26189} immature neutrophil "bands" in the center
{13643} immature neutrophil "bands"; there is also
a lymphocyte
{09809} Pap smear, trichomonas vaginitis, showing neutrophils
Viral infections, and certain unusual bacterial infections (typhoid, rickettsial disease) produce a NEUTROPENIA instead. You may see the same thing in an overwhelming infection.
Once acute inflammation has begun, there are four possible outcomes:
1. Complete RESOLUTION, i.e., there has been no damage to the connective tissue framework or non-recoverable cells of any part of the body.
2. Healing by SCARRING (see below)
3. ABSCESS formation. Pus in a confined space other than a natural body cavity is called an "abscess". As proteases continue to work on the proteins in the trapped fluid, the osmotic pressure within the abscess becomes greater and greater, causing it to swell ("ripen" -- ever had a pimple?) An abscess of course indicates that the body was not able to kill off all the bacteria (or whatever it was), but did contain them. As the pus builds up pressure, abscesses may find their way to a surface and rupture (as a pimple). In serious disease, usually you still have to drain pus.
4. Progression to CHRONIC INFLAMMATION (see below). This happens when, and only when, the neutrophils and their fast-acting molecular allies cannot remove the noxious agent.
MONONUCLEAR PHAGOCYTES
This is a generic term for blood monocytes and the cells to which they give rise. They are important in acute inflammation, as well as being a key element in chronic inflammation. Much of what you have just learned about neutrophils is equally applicable to monocytes.
{26440} monocyte in smear; most monocytes you see
will not have such good vacuoles
{26442} monocyte in smear
Like neutrophils, monocytes bear Fc and C3b receptors on their surfaces, in order to recognize opsonized materials for phagocytosis, and they will also engulf other kinds of particles. In addition to their famous role as scavengers, these cells ("all derived from the circulating blood monocyte") perform a host of other functions. Lone mononuclear phagocytes in the tissues are MACROPHAGES ("histiocytes", "dirt-bags", etc.), and may be fixed or mobile (but never so speedy as neutrophils).
Certain factors (notably GAMMA INTERFERON from T-cells) make macrophages angry ("activated"), increasing their ability to kill any organisms that they have devoured, and sometimes causing the macrophages themselves to adhere to form "granulomas" (see below). Other factors (notably TRANSFORMING GROWTH FACTOR β, also called "activin") de-activate them. Macrophages themselves generate a host of biological molecules.
* Macrophages that harbor many intracellular pathogens take on the appearance of "foam cells",
just as if they had eaten lots of free fat. We'll see these in
leprosy![]() ,
leishmaniasis
,
leishmaniasis![]() ,
rhinoscleroma,
malakoplakia, and xanthogranulomatous pyelonephritis.
,
rhinoscleroma,
malakoplakia, and xanthogranulomatous pyelonephritis.
ACUTE INFLAMMATORY MEDIATORS
You will find yourself overwhelmed if you try to learn all the effects of the chemical mediators of inflammation. This section includes items that are worth knowing for medical undergraduates.
VASOACTIVE AMINES include HISTAMINE and SEROTONIN, the classic mediators of immediate vascular permeability.
Histamine is immediately available from our mast cells. (Serotonin is found in rat mast cells.) These amines are released by trauma, cold, binding of antigen to the IgE on the mast cell surface, C3a and C5a, interleukin-1, and a host of histamine releasing factors from other white cells.
Histamine and serotonin are also released form our platelets ("the platelet release reaction").
Pharmacologists and clinicians: H1 receptors mediate the effects of histamine in inflammation.
THE COMPLEMENT COMPONENT SYSTEM is a group of 20 or so plasma proteins that are activated in cascades by the classic or alternate pathways (don't worry about the details now, just remember that the alternate pathway bypasses C4) or individually. Antigen-antibody complexes, dead tissue, and even plasmin activate ("fix") complement. Perennial test-bank items:
C3a and C5a ("the ANAPHYLATOXINS") increase vascular permeability, at least in part by releasing histamine from mast cells. C5a also liberates various chemotactic and noxious factors (notably arachidonic acid metabolites) from neutrophils and macrophages.
C3b is the great OPSONIN of the complement system.
C5b-9 is the MEMBRANE ATTACK COMPLEX, which punches holes in membranes of both friend and foe.
THE KININ SYSTEM is another group of proteins, which ultimately produce the nonapeptide bradykinin.
BRADYKININ increases vascular permeability, dilates blood vessels, contracts non-vascular smooth muscle, and causes pain. (Remember the last -- bee venom is largely bradykinin.)
* KALLIKREIN, another factor in the system, is chemotactic for neutrophils, and both activates and is activated by factor XII. Don't worry about the pathways of activation for these substances.
THE CLOTTING SYSTEM is a third system of proteins that you know. For now, just remember that activating the intrinsic pathway at its origin (factor XII) is one way to activate the kinin system, and that plasmin activates C3.
* Discussions of these cascades often make clotting factor XII ("Hageman factor") seem utterly central to the body's defenses. However, the real Mr. Hageman, who lacked the factor named for him, seemed none the worse for his deficiency -- he only learned late in life that his blood would not clot in a glass tube.
PROSTAGLANDINS: products of the cyclooxygenase pathway of arachidonic acid metabolism. (Review: The pathways in "Big Robbins", including the names of enzymes, are USMLE I pathology favorites.) Worth remembering:
THROMBOXANE A2 (TXA2), from platelets, aggregates platelets, constricts blood vessels (all the other prostaglandins are vasodilators, if that helps). Great for hemostasis.
* Thromboxane probably causes the cough due to the popular ACE-inhibitor antihypertensives (Lancet 350: 3, 1997). Not much clinically has come from thromboxane A synthesis inhibitors.
PROSTACYCLIN (PGI2), from the vessel wall, prevents platelet aggregation, dilates vessels. Great for whenever hemostasis is unnecessary.
PROSTAGLANDIN E2 (PGE2) is also a potent vasodilator (probably the most important one), greatly potentiates the ability of bradykinin to cause pain, and seems to be the local mediator of fever production for the hypothalamus. Both PGE and prostacyclin potentiate permeability-increasing and chemotactic mediators. All about prostaglandin E2: J. Immuno. 188: 21, 2012.
OTHER PROSTAGLANDINS exert a host of effects.
ASPIRIN, the NON-STEROIDAL ANTI-INFLAMMATORY DRUGS, and GLUCOCORTICOIDS inhibit cyclooxygenase, preventing the formation of the whole family.
LEUKOTRIENES: products of the lipooxygenase pathway of arachidonic acid metabolism. They are produced by all of the inflammatory cells except lymphocytes. Formerly called SRS or SLOW-REACTING SUBSTANCE(S). Review: J. Imm. 174: 589, 2005. Worth remembering:
LEUKOTRIENE C4 and its products D4, and E4 increase vascular permeability and constrict smooth muscle, and LEUKOTRIENE B4 makes polys adhere to endothelium and is a potent chemotactic agent.
Diets rich in omega-3 fatty acids prevent production of leukotrienes (and, to a lesser extent, prostaglandins). Leukotriene receptor antagonists are relatively new drugs that include "montelukast" and "zafirlukast". Leukotriene synthesis inhibitors (5-lipoxygenase inhibitors) are finally coming into use, especially for asthma ("zileuton").
Term: prostaglandins and leukotrienes are examples of AUTOCOIDS, i.e., short-range, locally-active hormones.
LYSOSOMAL CONSTITUENTS:
We have already seen that neutrophils release the contents of their granules during inflammation. For now, remember these neutrophils proteins:
Remember that monocytes produce acid hydrolases, collagenase, and elastase. Eosinophil specific granules contain several cationic proteins that seem to help fight the larger parasites.
Regardless of their sources, the proteases and free radicals released from inflammatory cells are can and do harm the body's own tissues. Most of this was studied in the 1990's and there's little current research.
The inflammatory response is often excessive. This is why, for example, it's probably best to put cold, rather than heat, on athletic and other minor injuries throughout the time they're healing. (* Tip from my best D.O. sports medicine consultant.)
The body has several proteins (notably α1-antitrypsin inhibitor, also known as "α1-protease inhibitor") to prevent proteases (from neutophils and elsewhere) from ruining our own tissues while we are still young. Remember that H2O2 and free radicals are also released from neutrophils and macrophages.
PLATELET ACTIVATING FACTOR, a small molecule, is generated on demand by various cells. Its various contributions to inflammation are only now being worked out (activates neutrophils and platelets, constricts smooth muscle, recruits and degranulates eosinophils), but the total effect is massive (Nature 374: 501, 1995). From time to time there's talk of platelet-activating factor inhibitors for therapy; no miracles.
NITRIC OXIDE: Dilates vessels locally (very fast), helps kill bacteria over the following several days, and has goodness-knows-how-many other effects: update J. Phys. Pharm. 54: 469, 2003.
CYTOKINES are polypeptide mediators made by lymphocytes ("lymphokines") and macrophages ("monokines"). Long familiar from immunology, it is now clear that they modulate the acute inflammatory response as well. Don't worry about the details in "Big Robbins".
The monokines INTERLEUKIN-1 and TUMOR NECROSIS FACTOR α ("cachectin" or "TNF-α") are key actors in the ACUTE PHASE REACTION, part of "just being sick" with an inflammatory illness.
During the acute phase reaction, there is somnolence, poor appetite, increased production and early release of neutrophils, and altered rates of hepatic synthesis of most of the major plasma proteins. Albumin and transferrin go down; α1-antitrypsin inhibitor, serum amyloid-associated protein, the complement components, fibrinogen, haptoglobin, and the atavistic (?) C-reactive protein go up.
* The serum chemistry changes of the acute phase reaction can be induced by the physical and psychological distress of military school hazing: Am. J. Clin. Nut. 53: 126, 1991. Interestingly, this preceded today's excitement over "low-grade systemic inflammation" as a big coronary risk factor, just like "stress" used to be (remember?)
* Interleukin 6 is up and down by the end of the first day after uncomplicated surgery. C-reactive protein is up and down by two days. Fibrinogen rises more slowly and is back down by 8 days or so.
The change in levels of plasma proteins is responsible for the INCREASED RED CELL SEDIMENTATION RATE, described by Hippocrates and still used to monitor the course of inflammation.
C-REACTIVE PROTEIN, long used in the clinical lab as a "marker for inflammation somewhere in the body", is now a subject of much interest.
* During the past decade, there has been a tremendous amount written about atherosclerosis and obesity as "inflammatory diseases", based almost entirely on the altered chemistry with elevated serum "inflammatory biomarkers". Even psychiatric depression is now discussed as if it were a systemic inflammatory disease (Am. J. Card. 96: 1016, 2005). The confusion is getting worse and worse -- exercise produces "a short-term inflammatory response", while being physically fit produces "a long-term anti-inflammatory effect".
* Maybe this is okay, since some of the mediators (notably TNF-alpha) are the same as in systemic diseases in which there is a lot of REAL inflammation, such as Crohn's disease and rheumatoid arthritis. Whether or not this use of the term "systemic inflammation" comes into common practice for any illness in which any inflammatory mediator is elevated in the bloodstream, I think it's proper to point out that this is not the historical meaning of the term "inflammation", and does not yet seem to be mainstream among pathologists. Without any disrespect, I'd suggest calling it "internist's inflammation", because the morphology is completely invisible to old-fashioned pathologists like me. We can talk about some of the cytokines familiar from localized inflammation being elevated in the blood of obese, lazy American children (Pediatrics 129: e1180, 2012), but do we really think of fat kids as inflammatory lesions like pimples, gonococcal urethritis, or gout? Further, "internist's inflammation" almost the opposite of what surgeons mean by "systemic inflammation" -- the catastrophic aberrations seen in the super-sick. Perhaps someone can think of a better term for the increase in "inflammatory" cytokines seen in couch potatoes and so forth -- the term "so-called low grade systemic inflammation" is used in Rheumatology 45: 944, 2006.
* The lymphokine LYMPHOTOXIN ("tumor necrosis factor β") has not received much attention in the past decade.
THE SYSTEMIC INFLAMMATORY RESPONSE SYNDROME ("total-body inflammation") represents toxicity from excessive production of the cytokines and/or other white-cell products.
We've mentioned the harm that an overly-excited inflammatory response can do to the body in severe illness. It's worth mentioning now the "usual suspects", since many are now the targets of today's biotech miracle drugs.
* Deltibant ("Bradycor"), an anti-bradykinin antagonist, was once promoted as improving survival in sepsis (JAMA 482: 277, 1997 -- didn't come into use) and for post-traumatic neuroprotection (J. Neurotrauma 16: 431, 1999 -- again, doesn't seem to have come into use.)
* Future clinicians wanting criteria: Make the call of SIRS when you have two of more of these and no other obvious explanation (Muscle Nerve 32: 140, 2005):
CHRONIC INFLAMMATION
THE HALLMARK OF CHRONIC INFLAMMATION IS INFILTRATION OF TISSUE WITH MONONUCLEAR INFLAMMATORY CELLS ("mononuclear cells", "round cells", i.e., MONOCYTES, LYMPHOCYTES, AND/OR PLASMA CELLS. Generally, good tissue has been (and is being) destroyed, and there will be some evidence of healing (scarring, fibroblast proliferation, angioblast proliferation).
{10973} lymphocytes and plasma cells in chronic inflammation
{10061} mostly lymphocytes;
{25397} autoimmune adrenalitis; low
power photo; many lymphocytes in the adrenal gland
{26430} small lymphocyte; notice that it is slightly
larger than the red cells
{26433} lymphocyte
{26436} lymphocytes, one resting, one a little bit turned-on (more cytoplasm,
more euchromatin)
{26412} plasma cell in a smear, top;
eccentrically-located soccer-ball (clockface) nucleus, abundant basophilic
cytoplasm, golgi pale spot
|
|
|
|
In clinically significant disease, we believe that the tissue macrophages are almost all recruited directly from the bloodstream monocytes. Plasma cells produce antibodies against the persistent antigen or the altered tissue components. Lymphocytes are likely to be present even where there is no involvement of the immune system.
Plasma cells appear in chronic inflammation as a result of T-helper cells activating B-lymphocytes. Interleukin 1 causes the B-cells to divide. The transformation into plasma cells is mediated (at least in part) by interleukin 4.
If IgE or worms are involved, you will probably see eosinophils. Their granules contains several alkaline ("basic") proteins that are noxious to worms.
The eosinophil proteins are now targets for specific therapies: J. Allerg. Clin. Imm. 113: 3, 2004.
{14708} eosinophil in smear
|
|
GRANULOMATOUS INFLAMMATION is a special kind of chronic
inflammation that occurs in the presence of indigestible material and/or cell-mediated immunity
("type IV hypersensitivity"; more about this in a few days). Ignore the definitions offered in textbooks.
A granuloma is an abnormal structure built from at least two activated macrophages adhering to one
another. Such macrophages are (confusingly) called EPITHELIOID CELLS. Granulomas serve to wall off
stuff (splinters, the caseous![]() debris of TB
debris of TB![]() , etc., etc.)
, etc., etc.)
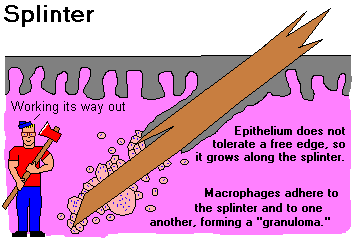
|
|
In the absence of a very large foreign body, a granuloma will almost always contain at least a few T-lymphocytes (though this is not absolutely mandatory).
The cells in a granuloma are activated by gamma-interferon (and/or α-TNF or whatever).
However, not all activated macrophages stick together. The current best candidate for "granuloma glue" is OSTEOPONTIN (Proc. Nat. Acad. Sci. 94: 6456, 1997; Science 287: 860, 2000; update Am. J. Path. 164: 567, 2004).
Whatever makes them the way they are, granulomas vanish as soon as the disease is effectively treated.
YOU MUST LEARN TO RECOGNIZE GRANULOMAS. Epithelioid cells have abundant pink cytoplasm, indistinct borders, and elongated, euchromatin-rich, reticulated nuclei oriented helter-skelter. My favorite gestalt: bluish-purple rice-crispies (nuclei) scattered on a frayed, pink tablecloth (cytoplasm).
{17629} granulomas in the lung
|
|
|
|
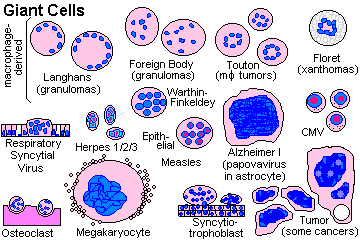
Granulomas can (but need not) contain syncytial GIANT CELLS (polykaryons). These fused clusters of epithelioid cells take a week to form. For our purposes, there are two kinds. LANGHANS GIANT CELLS have their nuclei arranged in a horseshoe around the edge, and FOREIGN BODY GIANT CELLS, with nuclei dispersed more or less evenly. The distinction is of no known significance.
|
|
{17628} epithelioid giant cell
The giant cells of granulomas occasionally contained altered cytoskeletal components in the shapes of stars, or ASTEROID BODIES. They are pretty, but of no known significance. Or you may see laminated calcified nuggets, called SCHAUMANN BODIES (* "conchoid bodies"), also of no known significance.
{25626} asteroid bodies in giant cells
{21428} granuloma with good asteroid body; this was a reaction to a jailhouse tattoo
|
|
|
|
|
The classic granulomatous diseases include tuberculosis![]() ,
tuberculoid leprosy
,
tuberculoid leprosy![]() ,
foreign body reactions
(* including the reactions to everything from sutures to
schistosome eggs
,
foreign body reactions
(* including the reactions to everything from sutures to
schistosome eggs![]() ), the deep fungal
infections, berylliosis, and the mysterious disease "sarcoidosis".
), the deep fungal
infections, berylliosis, and the mysterious disease "sarcoidosis".
* "Big Robbins" lists syphilis![]() (the granulomas, if any, are small and loose) and
silicosis (the granulomas, if any, are very fibrous).
(the granulomas, if any, are small and loose) and
silicosis (the granulomas, if any, are very fibrous).
|
{10958} tuberculosis, good caseous |
![]() TB granuloma
TB granuloma
Good caseous necrosis
WebPath Photo
* Future pathologists: Here is a reasonably complete catalogue of the granulomatous diseases.
GRANULOMAS WITH SUPPURATION (i.e., with pus in their centers; "stellate microabscesses") are typical of
those bacterial diseases with a propensity to involve lymph nodes. These are
lymphogranuloma
venereum![]() ,
cat scratch fever, brucellosis,
plague
,
cat scratch fever, brucellosis,
plague![]() ,
tularemia
,
tularemia![]() ,
glanders-melioidosis, listeria, campylobacter,
and
yersinia infection. East of Kansas City, don't forget
blastomycosis
,
glanders-melioidosis, listeria, campylobacter,
and
yersinia infection. East of Kansas City, don't forget
blastomycosis![]() .
.
{23386} lymphogranuloma venereum![]()
GRANULOMAS WITH CASEATION are typical of certain fungal infections (histoplasmosis![]() , blastomycosis,
and coccidioidomycosis
, blastomycosis,
and coccidioidomycosis![]() ,
as above) and of mycobacterial ("fungus-like bacteria") infections
(basically TB
,
as above) and of mycobacterial ("fungus-like bacteria") infections
(basically TB![]() ; also remember BCG bacillus,
and "atypical mycobacteria").
; also remember BCG bacillus,
and "atypical mycobacteria").
GRANULOMAS AROUND FOREIGN BODIES: aspirated food,
schistosome eggs![]() ,
toxocara, silicone injections,
splinters, sutures, windshield fragments, chalazions, ruptured epidermoid cysts, sea urchin spines,
mucus plugs in cystic fibrosis,
nitrogen bubbles ("pneumatosis"; "tissue emphysema"),
amyloidomas, dead aspergillus
,
toxocara, silicone injections,
splinters, sutures, windshield fragments, chalazions, ruptured epidermoid cysts, sea urchin spines,
mucus plugs in cystic fibrosis,
nitrogen bubbles ("pneumatosis"; "tissue emphysema"),
amyloidomas, dead aspergillus![]() fungi, dead filaria, ingrown hairs, talc in the lungs, metastatic
calcification bits, uric acid crystals (in longstanding gout, of course; these are "tophi"), sclerosing
lipogranuloma of the penis (J. Urol. 133: 1046, 1985, a fun article),
insect bites, "actinic
elastolytic granuloma of Mieschler" (a foreign body reaction to your
own elastic fibers), etc., etc.
fungi, dead filaria, ingrown hairs, talc in the lungs, metastatic
calcification bits, uric acid crystals (in longstanding gout, of course; these are "tophi"), sclerosing
lipogranuloma of the penis (J. Urol. 133: 1046, 1985, a fun article),
insect bites, "actinic
elastolytic granuloma of Mieschler" (a foreign body reaction to your
own elastic fibers), etc., etc.
Ruptured silicone breast implants produce aggregates of foamy macrophages (like macrophages loaded with lipid or mucin) but not good granulomas (Am. J. Clin. Path. 107: 236, 1997).
OTHER SOLID GRANULOMAS invite subclassification as immunologic diseases:
STRAIGHTFORWARD IMMUNE PROBLEMS: The organic pneumoconioses, berylliosis, zirconium disease (the infamous "armpit sarcoid", from zirconium-based deodorants), positive skin tests; certain of the mild hereditary immunodeficiency diseases (NEJM 358: 2030, 2008)
MORE ARCANE IMMUNE PROBLEMS: Wegener's granulomatosis (and its variants Churg-Strauss and lethal midline granuloma)
IMMUNOLOGIC REACTIONS TO TUMORS: Lennert's lymphoma, seminoma (both are often rich in granulomas); lymph nodes draining other cancers
MYSTERIOUS IMMUNE PROBLEMS: sarcoidosis, Crohn's disease, primary biliary cirrhosis, bronchocentric granulomatosis, the sarcoid / Crohn's-like inflammatory response seen near certain cancers
NEUTROPHIL DEFICIENCY SYNDROMES: notably "chronic granulomatous disease"
TOXOPLASMOSIS![]() (little loose granulomas)
and Q-FEVER
(little loose granulomas)
and Q-FEVER![]() (curious little "ring" granulomas) and
CUTANEOUS LEISHMANIASIS
(curious little "ring" granulomas) and
CUTANEOUS LEISHMANIASIS![]() ("foamy
granulomas", present if immune response is good). Baboon amoebas (don't worry
about them just now: Lancet 362: 220, 2004), and CNS amoebas in the immunocompromised
("foamy
granulomas", present if immune response is good). Baboon amoebas (don't worry
about them just now: Lancet 362: 220, 2004), and CNS amoebas in the immunocompromised
HIV ENCEPHALITIS presents groups of giant cells, the result of macrophages recognizing HIV protein on each others' surfaces
SCARRING means LAYING-DOWN OF DENSE (type I) COLLAGEN IN CHRONIC INFLAMMATION AND/OR WOUND HEALING (see below; brain makes its scars out of glial filaments instead). Usually, when there is chronic inflammation of any time, some dense collagenous scar gets laid down.
Right now, TRANSFORMING GROWTH FACTOR β gets most of the credit (blame) for causing fibrosis in chronic inflammation. Interleukin 1, from macrophages, is also a potent activator of fibroblasts. This probably accounts for part of the scarring in chronic inflammatory diseases.
Actually, the whole subject of fibrosis (especially situations in which there's too much) has become enormously complicated as we've learned more about mediators. Tomorrow's treatments will come from this knowledge. Update NEJM 372: 1138, 2015.
With the new emphasis on following the various types of inflammatory cells, it's now clear that some types of inflammation are the sequelae of injurious agents that are gone. The best-established is the ongoing inflammation in the lungs of ex-smokers (J. Immuno. 186: 5457, 2011; Am. J. Med. 125: 1162, 2012). Don't be surprised if a variety of "mysterious chronic inflammatory illnesses" turn out to belong in this category as well.
An ULCER (* "ulceration", for those who prefer nouns made from verbs made from nouns) forms when necrosis has involved a body surface and a portion of it is sloughed. Further, there must be necrosis of both the epithelium and at least some of the underlying connective tissue.
|
|
Australian Pathology Museum High-tech gross photos
|
|
|
Note that any definition of an ulcer must exclude paper cuts (i.e., breaks in surfaces without necrosis) and unroofed friction blisters (i.e., loss of epithelium without loss of connective tissue.)
Ulcers are discussed here by "Big Robbins" because they are always inflamed.
We've seen pictures of ulcers when we discussed necrosis. Please note that the familiar, banal DECUBITUS ULCER of pressure points results from ischemic necrosis. (Do you understand how?)
A little-known fact is that decubiti of the colonic and rectal mucosa from fecal impaction are common, and can be the portal of entry for bacteria. These are called "stercoral" or "stercoraceous" ulcers, and they can easily kill a person.
{10461} duodenal ulcer (stomach is at top)
{10471} stomach ulcer (esophagus at right,
duodenum at left)
{53543} stomach ulcer (a section has already
been taken by the pathologist)
{10811} stomach ulcer, side view of a section through the crater;
see how the ulcer has penetrated through the muscularis propria and
only scar prevents perforation)
{11651} bad foot ulcer
{15560} bleeding stomach ulcer (arrow marks bleeding site)
{48177} diabetic ulcer
A PSEUDOMEMBRANE results when the upper portion of a mucosal surface undergoes necrosis, freeing fibrinogen from vessels that then clots along the surface. A pseudomembrane is actually a very large, very shallow ulcer. The best pseudomembranes include secretory product from the underlying glands as well.
{10529} pseudomembranous enterocolitis
|
|
NOTE: Very confusing to students is sloppy use of the term "chronic inflammation" for scarring left over from acute inflammation that resolved long ago. "Chronic pyelonephritis", "chronic pancreatitis", and "chronic pericarditis" are generally misnomers -- there's little or no inflammation left, just scar.
* Ignore old-fashioned discussions of "serous", "fibrinous", "hemorrhagic", "suppurative" and "purulent" inflammation. Remember that really severe inflammation will allow fibrinogen out of the vessels. CATARRH is an archaic word for an exudate, or for heavy secretion from an inflamed mucous membrane.
* In the future, look for much more about mediators in inflammation produced by epithelium and fibroblasts, especially as causes of "idiopathic" diseases in which chronic inflammation figures prominently.
REGENERATION
|
|
|
Inflammation is said to RESOLVE when no structural cells have been lost after the inflammatory process is complete and phagocytosis has cleaned up the area. When the tissue has been damaged during the inflammatory process or in other ways, but the body itself is still alive, the tissue will either REGENERATE or BE REPAIRED ("organized") BY FIBROUS TISSUE. If none of the latter is required, the word "resolution" is also appropriate. If any repair by fibrous tissue occurs, there will be a SCAR. (Depending on the site, scar tissue may be called "cicatrix", "fibrosis", "adhesions", "gliosis", "fibroplasia", etc., etc.)
| We rank cells according to their ability to regenerate: |

|
LABILE CELLS ("continuous replicators") are constantly replenishing their neighbors that have died or been shed. Examples include the epithelium of skin, mucous membranes, oviducts, ducts; urothelium; endometrium; seminiferous tubules; bone marrow; lymphoid tissue.
Probably these cells would "like to" proliferate all the time, but are stopped by "contact inhibition" by their neighbors. More about this arcane subject when we talk about cancer....
Epidermis can regenerate from the skin adnexal structures (hair follicles, sebaceous glands, sweat glands), enabling full removal of epidermis as for a skin graft.
STABLE CELLS ("discontinuous replicators") can proliferate rapidly in response to need, especially when required to replace lost neighbors. These include all glandular parenchymal cells, as well as fibroblasts, endothelial cells (cuboidal, and called "angioblasts", when they are healing), and osteoblasts.
Skeletal muscle, cartilage and tendon heal rather poorly, since nothing will restore their specialized structure. Smooth muscle cells regenerate poorly.
The champion healer is the liver. The parenchymal cells seem to have unlimited capacity to regenerate. And it's almost impossible to destroy its connective tissue framework in the short-term.
PERMANENT CELLS ("non-replicators") have very limited ability to undergo mitosis or be replenished after birth. These cells include glia, neurons, and cardiac (non-failing heart).
* In animals models, neurons can reappear from stem cell progenitors (even in response to SSRI's -- see Science 301: 757, 2003).
* The regenerative ability of the myocardial cell: Lancet 363: 1306, 2004. Maybe someday this will be clinically useful.
Obviously, cells will not regenerate if there is inadequate blood supply, inadequate nutrition, or complete destruction of their connective tissue framework.
* Someone will tell you, "The more specialized the tissue, the less its powers of regeneration." This isn't true. Liver regenerates, and belly button doesn't.
|
|
|
|
|
REPAIR BY CONNECTIVE TISSUE
And the world will be better for this, that one man scorned and covered with scars still strove with his last ounce of courage to reach the unreachable stars.
The history of a soldier's wound beguiles the pain of it.
|
 Shakespeare's Romeo & Juliet |
You already know the "law of epithelium" -- it will not tolerate a free edge. In other words, an epithelial cell without a neighbor will divide to replace it. This is a rapid process, and re-epithelialization happens as long as there is any "free edge" nearby.
A few hours after injury, there is already evidence of connective tissue repair. Fibroblasts become active and begin to proliferate, and buds ("angioblasts") sprout from the damaged capillaries. Of course, the cells will show lots of euchromatin, large nucleoli, and abundant basophilic cytoplasm. Typically, both kinds of cells invade the fibrin meshwork created during the injury and inflammatory response.
The fibroblasts produce ground substance, fibronectin, and type III collagen; later they will produce type I collagen for the mature scar.
The young vessels are leaky, so healing wounds are edematous both grossly and microscopically. The fibroblasts lay down collagen and proteoglycans ("ground substance"), and some acquire contractile elements composed of cytoskeletal components as in smooth muscle ("myofibroblasts"). Of course, there are plenty of macrophages (to keep the new tissue clean) and mast cells. The new tissue is called GRANULATION TISSUE ("immature scar", etc.), and the fibrin meshwork is said to be undergoing ORGANIZATION. (* Don't worry about the several other plasma-derived proteins that are also present in the area after injury and have to do with healing.) You've seen granulation tissue -- it was moist, red, jelly-like stuff under the scab that you picked off too soon.
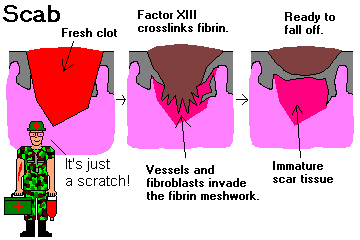
|
|
|
|
Exactly how new blood vessels loop into vascularizing tissue is still under much study. Update Nat. Med. 15: 608 & 657, 2009.
* You may run into granulation tissue that doesn't mature; depending on its location, you may call it an "inflammatory pseudotumor", or whatever.
If everything goes well, eventually there is sufficient collagen to fill the gap (type I replaces the type III originally laid down in the granulation tissue), most of the capillaries are reabsorbed, the fibroblasts revert to a resting mode, and finally the myofibroblasts contract.
Especially where there has only been chronic inflammation, you can also see dense collagen production, which of course also counts as scar tissue. This is done by fibroblasts on the instructions of macrophages.
{12707} granulation tissue
{17606} granulation tissue in healing ulcer
{17607} granulation tissue in healing ulcer
{17608} granulation tissue
{17609} granulation tissue
{17610} granulation tissue in healing ulcer
{17611} granulation tissue in healing ulcer
|
|
HEALING BY PRIMARY INTENTION
A well-approximated surgical wound is the ideal situation for wound healing. Since the edges are close together and held tight by sutures and fibrin, and there is little necrosis and hopefully no infection, the healing is by PRIMARY UNION or FIRST INTENTION.
Timetable for "the best possible wound" (i.e., a clean, protected one with edges apposed, in a well-nourished patient with good blood vessels):
minutes: Fibrinogen from the severed vessels is activated via one or the other arms of the clotting cascade, forms a meshwork, and stops the bleeding. The meshwork also contains platelets.
24 hours: Polys have entered the fibrin meshwork. Epithelial cells are regenerating from the edges of the wound surface, etc.
3 days: The fibrin meshwork is extensively invaded by macrophages. Granulation tissue is appearing at the edges of the incisions. A thin layer of epithelial cells now covers the wound surface.
5 days: Granulation tissue fills the entire wound, and there is easy-to-see collagen.
2 weeks: Fibroblasts continue to multiply, and collagen continues to accumulate.
4 weeks: The overlying epidermis is now normal, though it will not re-grow adnexal structures. Capillary involution and scar contraction is well underway, and the red scar is turning white. The wound is still growing stronger, though it will never have the tensile strength of uninjured tissue (sorry).
{08210} skin scar, nicely healed
HEALING BY SECONDARY INTENTION
Most wounds do not conform to the above ideal. There is a larger fibrin meshwork (a SCAB, rich in red cells -- now brown because of oxidation of the heme pigment), more inflammation, possibly infection, more granulation tissue, and more spectacular wound contraction (up to 90-95% of the original surface area.)
When epidermis grows underneath some of the fibrin meshwork, the edges of the scab loosen. When re-epithelialization is complete, the scab falls off.
As surface epithelium grows into crevices (i.e., down suture tracks, etc.), it excites excessive fibroblastic activity. This is why there's more scarring where the sutures were.
Scarring by secondary intention always produces some deformity.
The weave of collagen in the final scar (primary or secondary intention) is never the same as in the surrounding connective tissue.
Sometimes the granulation tissue undergoes striking proliferation beyond the wound margins. This is called EXUBERANT GRANULATIONS by physicians and "proud flesh" by the public. (You'll excise it.)
More intractable are KELOIDS, (literally "crab claws") disfiguring scars with excessive collagen production, seen primarily in darkly-pigmented people.
* Treating keloids usually involves re-excision and injection of the surgical bed with glucocorticoid and/or interferon and/or administration of surface ionizing radiation.
{12805} "keloids", gross
{12807} "keloid", elbow
{24487} keloid, after a burn
{40368} "keloid", ear lobe
{25527} keloid, histology
{26432} keloid, histology
{26438} keloid, histology ("glassy fibers")
|
|
Even worse than ugly surface scars are acquired deformities of the cardiac valves, and scars that compress or plug the lumens of hollow organs.
If a scar is subjected to continual strain, the wound will stretch. Incisional hernias are the best examples of this phenomenon.
Other mishaps may occur.
Pigment in a wound is likely to stay in the macrophages. Hemosiderin may persist for years in a scar, especially if the person already has a high total-body iron burden.
Fragments of epidermis trapped in a healed wound may grow into spheres "with the skin-side inside" -- the familiar "epidermal inclusion cysts" ("sebaceous cysts", etc.)
Attempts by severed sensory nerves to grow back into wounded tissue may produce painful "traumatic neuromas".
NOTE: The wall of an abscess is, of course, granulation tissue.
WHAT MAKES WOUND HEALING HAPPEN?
As for inflammation, GROWTH FACTORS for wound healing are continually being discovered. "Big Robbins" lists the seven growth factors that seem to direct the production of granulation tissue. You should recognize PLATELET-DERIVED GROWTH FACTOR as a key to fibroblast activation and fibrogenesis, and recognize the names of the others ("epidermal growth factor", "fibroblast growth factor", "transforming growth factors α and β", interleukin 1, and TNF/cachectin.) ANGIOGENESIS is obviously central to wound healing, but the molecules are still getting discovered.
A PROVISIONAL MATRIX forms in the injured area from molecules derived from the plasma -- even if fibrin is not present, hyaluronan, tenascin and fibronectin are laid down and can be scaffolding for granulation tissue and healing.
FIBRIN itself seems to attract inflammatory cells, fibroblasts, and angioblasts. CONTACT INHIBITION and crowding seem to put the brakes on the process. Material in "Big Robbins" on cell-cell and cell-matrix interactions are still experimental. Now is a good time to read up on "integrins" in your biochemistry book; you'll prescribe such medicines as natalizumab (α4 integrin antagonist that has been found to be useful in Crohn's disease and multiple sclerosis).
FACTORS MODIFYING INFLAMMATION AND REPAIR
Despite conventional wisdom, AGE does not exert much effect on inflammation or wound healing.
ADEQUATE NUTRITION is needed for good wound healing. Protein is needed for collagen synthesis, and vitamin C for hydroxylation of the proline and lysine in collagen. Several enzymes required for wound healing are zinc-based. Some surgeons supplement some or all of these nutrients for their post-operative patients.
INADEQUATE BLOOD SUPPLY greatly interferes with both inflammation and healing.
WOUND INFECTION interferes with timely wound healing. Foreign bodies (dirt, sutures, others) in a wound are a tremendous aid to bacteria in causing infections, as the bugs can cling to the surfaces and thus escape phagocytosis.
We do not know exactly how GLUCOCORTICOIDS interfere with wound healing, but the effect is potent. (For starters, they inhibit the migration of fibroblasts into fibrin meshworks.)
Future surgical clerks: Here are some names for surgical operations!
-tomy: The surgeon cut something.
-ectomy: The surgeon cut something out.
-ostomy: The surgeon cut something to make a mouth. If one organ is named, the mouth opened to the outside of the patient. If two organs are named, the mouth connected two organs.
-plasty: The surgeon changed the shape of an organ.
-pexy: The surgeon moved the organ to the right place.
-rraphy: The surgeon sewed something up.
-desis: The surgeon made two things stick to one another.
RULES OF THUMB:
In infections by the common bacteria
(staphylococci![]() ,
streptococci
,
streptococci![]() ,
gram-negative rods or cocci), the
predominant cell in the inflammatory infiltrate (and in the circulating
blood of the infection is systemic) is the NEUTROPHIL.
,
gram-negative rods or cocci), the
predominant cell in the inflammatory infiltrate (and in the circulating
blood of the infection is systemic) is the NEUTROPHIL.
In viral infections and autoimmune diseases, the predominant cell in the inflammatory infiltrate is the LYMPHOCYTE. In viral illnesses, there may be a brief increase or decrease in the circulating lymphocytes.
There might be some neutrophils early-on in the tissues injured by viral infections.
Whooping cough (pertussis) produces a spectacular increase in circulating lymphocytes.
In the spirochetal diseases (syphilis![]() and Lyme
disease
and Lyme
disease![]() ),
the predominant cell in the inflammatory infiltrate is the PLASMA CELL
along with plasmacytoid lymphocytes.
),
the predominant cell in the inflammatory infiltrate is the PLASMA CELL
along with plasmacytoid lymphocytes.
In typhoid fever, SARS, tuberculosis, and fungal infections (except some hyphal forms and
candidiasis![]() ), there's often
an increase in circulating monocytes (helps make the diagnosis of typhoid
especially) and the predominant cell in the
inflammatory infiltrate is the MONOCYTE / MACROPHAGE
/ HISTIOCYTE / EPITHELIOID CELL.
), there's often
an increase in circulating monocytes (helps make the diagnosis of typhoid
especially) and the predominant cell in the
inflammatory infiltrate is the MONOCYTE / MACROPHAGE
/ HISTIOCYTE / EPITHELIOID CELL.
* As above, in lymphogranuloma
venereum![]() ,
cat scratch fever, brucellosis,
plague
,
cat scratch fever, brucellosis,
plague![]() ,
tularemia
,
tularemia![]() ,
glanders-melioidosis, and yersinia infection, there will be a plentiful mix of neutrophils and
epithelioid histiocytes.
,
glanders-melioidosis, and yersinia infection, there will be a plentiful mix of neutrophils and
epithelioid histiocytes.
In infections caused by metazoan parasites (i.e., worms), and in Hodgkin's disease, and in many nonspecific inflammations in the gut, a predominant cell in the inflammatory infiltrate is the EOSINOPHIL. In Hodgkin's and severe worm infections, eosinophils are likely to be elevated in the blood as well.
But depending on the agent and the host, there may not be ANY inflammatory reaction!
![]() Viral lung infection
Viral lung infection
Lymphocytes
WebPath Photo
TYPES OF PAIN
If you have missed this so far in your medical education, learn it now.
In questioning people about their pain, you ask:
ACHING PAIN: Probably periosteum, tooth, dura, or some circuit inside your own brain is involved
BURNING PAIN: Either (1) the integrity of a mucosal surface has been breached, or (2) the nerve or its immediate environment has been damaged.
CRAMPY PAIN (gas, labor, kidney stones): A hollow organ is being distended
STABBING PAIN ("lancinating") (pleuritis, pericarditis, peritonitis): If you haven't really been stabbed, then one of your serosal membranes is hurting.
NOT REALLY ANY OF THESE: ischemia, common inflammation (everything from bee-sting to
plague![]() )
)
Buttercup: You mock my pain.
Westley: Life is pain, Highness. Anyone who says differently is selling something.
-- The Princess Bride
* If you want to get good at looking at pictures, and you have extra time, enjoy these pathology
pictures of inflamed tissue now:
{08121} polyp, inflammatory
Romeo wanted to be loved.
{09322} polyp, inflammatory fibroid
{09806} inflammation, non-specific changes on a pap smear; look at nice neutrophils and the nuclei of the epithelial cells with
some big nucleoli
{09822} polyp, inflammatory polyp in stomach
{10058} inflammation, mixed acute and chronic
{10061} inflammation, chronic
{10106} inflammation, granulomatous
{10229} inflammation, granulomatous
{10529} inflammation, pseudomembranous colon
{10973} inflammation, chronic soft tissue
{12186} hypopigmentation, post-inflammatory
{12207} hyperpigmentation, post-inflammatory
{13153} inflammation, submandibular gland
{14350} dermatomyositis, severe inflammation
{14352} dermatomyositis, severe inflammation
{14432} polymyositis, acute inflammatory
{14433} polymyositis, acute inflammatory
{14702} polymorphonuclear leukocyte, normal
{14703} polymorphonuclear leukocyte, normal
{14704} polymorphonuclear leukocyte, normal
{14705} polymorphonuclear leukocyte, normal
{14706} polymorphonuclear leukocyte, normal
{14707} polymorphonuclear leukocyte, normal
{14734} polymorphonuclear leukocyte & lymphocyte
{14735} polymorphonuclear leukocyte & lymphocyte
{16061} chronic inflammation, liver
{16188} polymorphonuclear leukocyte
{16194} polymorphonuclear leukocyte
{16365} post inflammatory ossification
{16366} post inflammatory ossification
{16367} post inflammatory ossification
{16368} post inflammatory ossification
{16369} post inflammatory ossification
{16370} post inflammatory ossification
{16767} inflammation, glomerulus
{16771} neutrophil stuck to glomerular basement membrane
{17406} necrosis, fibrinoid inflammatory artery
{17612} inflammation, chronic acute skin (no normal tissue here)
{17613} inflammation, chronic
{17614} inflammation, chronic
{17615} inflammation, chronic
{17616} inflammation, chronic in skin
{17618} inflammation, chronic and acute
{17622} inflammation, lung
{17630} inflammation, chronic granulomatous lung
{20782} polymorphonuclear leukocyte, normal
{24652} inflammation, perivascular muscle
{24680} polyp, inflammatory colon
{25041} atypia, inflammatory
{25049} atypia, inflammatory
{25117} lymphogranuloma venereum![]()
{25198} fibrous pseudotumor
{25776} single cell arrangement
{25869} inflammation, pap smear
{25897} inflammatory atypia - mild
{26001} lymphocyte
{26007} lymphocyte, chronic inflammation
{26107} inflammation, chronic
{26109} inflammation, acute and bacteria
{26187} promyelocyte and neutrophil segment
{26188} promyelocyte and neutrophil segment
{26189} neutrophil, band
{26190} neutrophil, band
{26191} neutrophil segment
{26192} neutrophil segment
{26193} neutrophil segment
{26194} neutrophil segment
{26201} neutrophil, band and metamyelocyte
{26202} neutrophil, band and metamyelocyte
{26211} myelocyte, neutrophil
{26212} myelocyte, neutrophil
{26214} neutrophil, band
{26215} neutrophil, band
{26230} polymorphonuclear leukocyte, normal
{29997} title slide
{30268} inflammation, brain
{30269} inflammation, brain
{32242} leishmaniasis![]() in skin inflammation
in skin inflammation
{32287} brugia microfilaria with neutrophils
{34748} inflammation, vaginal smear
{38377} endarteritis, inflammatory
{39832} pelvic inflammatory disease
{40392} hydrocele; inflamed
{46186} polyarteritis
{46192} vasculitis
{46194} candida![]() pseudohyphae (silver)
pseudohyphae (silver)
{46195} candida![]() pseudohyphae (H&E)
pseudohyphae (H&E)
{46199} toxoplasma![]()
{46204} stenosis rheumatic mitral
{46213} pneumococcal septicemia
{46214} listeria
{46215} pyomyositis
{46218} tuberculoma
{46219} meningitis, tuberculous![]()
{46221} trichinosis
{46222} hepatitis herpes simplex![]()
{46223} hepatitis herpes simplex
{46224} nocardia![]()
{46225} coccidioidomycosis![]()
{46226} histoplasma capsulatum![]()
{46227} histoplasma capsulatum![]()
{46228} histoplasma capsulatum![]()
{46229} histoplasma duboisii
{46230} aspergillus fumigatus![]()
{46233} candida albicans![]()
{46234} cryptococcus![]()
{46235} trichomonas mentagrophytes
{46236} aspergillus flavus![]()
{46237} actinomyces israeli![]()
{46238} nocardia![]()
{46239} nocardia![]()
{46240} Microsporum audouini
{46241} leishmaniasis![]() (cutaneous)
(cutaneous)
{46242} leishmaniasis![]() (cutaneous)
(cutaneous)
{46243} leishmaniasis![]() (kala azar)
(kala azar)
{46247} malaria![]() p. falciparum
p. falciparum
{46248} malaria p. falciparum
{46249} malaria cerebral
{46251} stenosis rheumatic mitral
{46259} aspergillus septicemia![]()
{46262} bronchiectasis and tuberculosis
{46263} asthma
{46268} Crohn's disease
{46289} herpes simplex![]()
{46300} lupus band
{46301} dermatitis herpetiformis iga
{46305} rheumatic fever
{46310} pyelonephritis acute
{46311} pyelonephritis xanthrogranulomatous
{46317} Berger's disease (h&e)
{46318} Berger's disease (iga)
{46390} spondylitis ankylosing
{46391} rheumatoid arthritis synovium
{46392} rheumatoid arthritis
{46396} polyarteritis nodosa
{46441} Crohn's disease
{46443} amebiasis
{46466} carbon tetrachloride injury 24 hours
{46467} radiation injury
{46469} pneumonia cytomegalovirus
{46472} mast cell
{46473} mast cell degranulation
{46474} macrophage migration inhibition
{46475} Chagas' disease
{46476} Chagas' disease
{46477} Chagas' disease
{46478} cysticercus cellulosae
{46480} rheumatic valvulitis
{46481} rheumatic heart disease anitschkow myocyte
{46484} Crohn's disease
{46486} diarrhea (30 mins.)
{46491} treponema (Levaditi)
{46492} candida![]() septicemia
septicemia
{46493} echinococcus
{46495} trichuris
{46496} filaria
{46497} babesia
{46498} trichomonas vaginalis
{46505} tuberculosis
{46532} phlebitis pericentral
{46543}
hyaline droplets
{46548}
fibrin
{46551}
mast cell degranulating
{47613}
tooth pulp, inflamed
{47614}
tooth pulp, inflamed
{47621}
tooth pulp, inflamed
{47622}
tooth pulp, inflamed
{53544}
inflammation, necrotizing wall
{53545}
inflammation, necrotizing wall
{53546}
inflammation, chronic after inflammation
{06190}
schistosomiasis ![]() egg shell in granuloma with giant cells
egg shell in granuloma with giant cells
{07061}
trauma vascular obstruction
{07062}
trauma vascular obstruction
{07152}
trauma vascular obstruction
{09120}
granuloma
{09246}
thyroiditis, granulomatous
{09247}
thyroiditis, granulomatous
{09854}
Q-fever hepatitis, ring granuloma![]()
{10106}
inflammation, granulomatous
{10229}
inflammation, granulomatous
{10958}
granuloma, lymph node caseous![]()
{10964}
granuloma, lymph node caseous
{10976}
granulomas, tonsil
{10979}
granulomas, tonsil
{10982}
granulomas, tonsil
{11423}
granuloma, lung fibrocalcific
{11541}
pyelonephritis, xanthogranulomatous
{11659}
granuloma, lung necrotizing
{11990}
Wegener's granulomatosis, necrotic nodule
{11991}
Wegener's granulomatosis, pulmonary
{12225}
granuloma, pyogenic
{12226}
granuloma annulare
{12356}
erosion, granuloma inguinale
{12783}
granuloma, pyogenic
{12784}
granuloma, pyogenic
{12786}
granuloma, pyogenic
{12787}
granuloma, pyogenic
{12789}
granuloma, pyogenic
{12790}
granuloma, pyogenic
{13053}
granuloma annulare
{13054}
granuloma annulare
{13056}
granuloma annulare
{13180}
granuloma, larynx
{13185}
granuloma, vocal cord
{13364}
histoplasmosis![]() , lung; granuloma
, lung; granuloma
{13365}
histoplasmosis![]() , lung; granuloma
, lung; granuloma
{13670}
granuloma, lymph node
{14427}
sarcoid, diffuse granulomatous myositis
{15376}
granuloma, lymph node
{15379}
granuloma with necrosis DF-2
{15671}
granuloma, eosinophilic
{15672}
granuloma, eosinophilic
{15746}
schistosoma eggs![]()
{15757}
schistosoma![]()
{17629}
inflammation, granulomatous lung
{17630}
inflammation, chronic granulomatous lung
{17631}
granuloma, lung
{19512}
colitis, granulomatous
{20125}
lipogranulomatosis, farber's
{20127}
lipogranulomatosis, farber's
{20206}
granulomas, spleen
{21285}
granuloma, peripheral giant cell
{21286}
granuloma, peripheral giant cell
{21294}
granuloma, peripheral giant cell
{21295}
granuloma, peripheral giant cell
{21296}
granuloma, peripheral giant cell
{21297}
granuloma, peripheral giant cell
{21298}
granuloma, peripheral giant cell
{21303}
granuloma, traumatic
{21367}
granuloma, central giant cell
{21497}
granuloma, central giant cell
{21531}
granuloma, peripheral giant cell
{21532}
granuloma, peripheral giant cell
{21533}
granuloma, peripheral giant cell
{21534}
granuloma, peripheral giant cell
{21535}
granuloma, peripheral giant cell
{21541}
granuloma, traumatic
{21542}
granuloma, traumatic
{21543}
granuloma, traumatic
{21545}
granuloma, traumatic
{21546}
granuloma, traumatic
{21584}
granuloma, pyogenic
{21585}
granuloma, pyogenic
{21586}
granuloma, pyogenic
{21587}
granuloma, pyogenic
{21602}
epulis, congenital=giant cell granuloma
{21603}
epulis, congenital=giant cell granuloma
{21604}
epulis, congenital=giant cell granuloma
{21605}
granuloma, peripheral giant cell
{21606}
granuloma, peripheral giant cell
{21607}
granuloma, peripheral giant cell
{21710}
granuloma, pyogenic
{21721}
granuloma, peripheral giant cell
{21777}
granuloma, peripheral giant cell
{21880}
pyogenic granuloma, conjunctiva
{21882}
pyogenic granuloma, conjunctiva
{21883}
pyogenic granuloma, conjunctiva
{23386}
lymphogranuloma venereum![]()
{23422}
sarcoidosis, granulomas
{23425}
lymphogranuloma venereum![]()
{23968}
prostatitis, granulomatous
{23999}
granuloma, post surgical
{24005}
granuloma, BCG
{24006}
granuloma, BCG
{24044}
pyelonephritis, xanthogranulomatous
{24045}
pyelonephritis, xanthogranulomatous
{24720}
thyroiditis, granulomatous
{24721}
thyroiditis, granulomatous
{24760}
granuloma, cholesterol tendon
{24852}
granulomatosis, Wegener's kidney
{24886}
xanthogranuloma, juvenile
{24918}
granuloma annulare
{25089}
granuloma inguinale
{25090}
granuloma inguinale, silver stain
{25117}
lymphogranuloma venereum![]()
{25140}
epididymitis with sperm granulomas
{25141}
epididymitis with tubal rupture
{25142}
orchitis, granulomatous
{25143}
orchitis, granulomatous
{25196}
sperm granuloma
{25218}
prostatitis, granulomatous
{25220}
tuberculosis
{25340}
pyelonephritis, xanthogranulomatous
{27503}
berylliosis, granulomas
{27667}
coccidioides, granuloma![]()
{27672}
coccidioides, granuloma![]()
{29997}
title slide
{29998}
inflammatory granulomatous syndromes
{29999}
inflammatory granulomatous syndromes
{30000}
inflammatory granulomatous syndromes
{30001}
inflammatory granulomatous syndromes
{31996}
aspergillus![]() granuloma, brain
granuloma, brain
{32215}
schistosoma mansoni egg granuloma![]()
{32226}
pyogenic granuloma in skin
{32227}
schistosoma mansoni egg granuloma![]()
{32251}
schistosoma egg granuloma![]()
{32277}
schistosomal granuloma![]()
{32311}
schistosoma japonicum![]() granuloma fibrosis
granuloma fibrosis
{32321}
granuloma, noncaseating
{32324}
granuloma, noncaseating
{32346}
schistosoma japonicum![]() granuloma, mouse
granuloma, mouse
{34147}
granuloma, eosinophilic
{34150}
granuloma, eosinophilic
{35549}
beryllium, granuloma
{35552}
beryllium, granuloma
{36790}
granuloma, eosinophilic
{36793}
granuloma, eosinophilic
{37462}
granuloma, cryptococcus![]()
{38881}
pyelonephritis, xanthogranulomatous
{39496}
granulomatosis, Wegener's
{39497}
granulomatosis, Wegener's
{39543}
granulomatosis, Wegener's
{39573}
pyelonephritis, xanthogranulomatous
{39574}
pyelonephritis, xanthogranulomatous
{39575}
pyelonephritis, xanthogranulomatous
{39879}
granuloma, fungal
{39880}
granuloma, fungal
{40073}
xanthogranuloma
{40120}
salpingitis, xanthogranulomatous
{40351}
granuloma
{40352}
granuloma
{40520}
granulomatosis, Wegener's
{40521}
granulomatosis, Wegener's
{40522}
granulomatosis, Wegener's
{40523}
granulomatosis, Wegener's
{40524}
granulomatosis, Wegener's
{40525}
granulomatosis, Wegener's
{40526}
granulomatosis, Wegener's
{40527}
granulomatosis, Wegener's
{40568}
granuloma, giant cell
{40569}
granuloma, giant cell
{40570}
granuloma, giant cell
{40571}
granuloma, giant cell
{40662}
granuloma, eosinophilic
{40663}
granuloma, eosinophilic
{40664}
granuloma, eosinophilic
{46311}
pyelonephritis xanthrogranulomatous
{49307}
pyelonephritis, xanthogranulomatous
{49350}
lipogranuloma from ruptured breast implant
{49459}
thyroiditis, granulomatous
Don Quixote wanted to find
meaning.
Tristram Shandy wanted to find
an answer for death.
All are great reads. Watch these themes
as you go through your medical education and practice.
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about the essential processes of disease to consult these standard textbooks.
In my notes, the most helpful current journal references are embedded in the text. Students using these during lecture strongly prefer this. And because the site is constantly being updated, numbered endnotes would be unmanageable. What's available online, and for whom, is always changing. Most public libraries will be happy to help you get an article that you need. Good luck on your own searches, and again, if there is any way in which I can help you, please contact me at scalpel_blade@yahoo.com. No texting or chat messages, please. Ordinary e-mails are welcome. Health and friendship!


| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |


 Inflammation and Tissue Repair
Inflammation and Tissue Repair
