Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com
No texting or chat messages, please. Ordinary e-mails are welcome.

|

|
 |
 |
 |
 |
|
verify here. |
This website collects no information. If you e-mail me, neither your e-mail address nor any other information will ever be passed on to any third party, unless required by law.
This page was last modified January 1, 2016.
I have no sponsors and do not host paid advertisements. All external links are provided freely to sites that I believe my visitors will find helpful.
Cyberfriends: The help you're looking for is probably here.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected. No texting or chat messages, please. Ordinary e-mails are welcome.
 I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site
at www.afraidtoask.com.
I am active in HealthTap,
which provides free medical guidance from your cell phone.
There is also a fee site
at www.afraidtoask.com.
 If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |

|
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource.
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
 My team:
My team:
pathology.org -- my cyberfriends, great for current news and browsing for the general public
EnjoyPath -- a great resource for everyone, from beginning medical students to pathologists with years of experience
Medmark Pathology -- massive listing of pathology sites
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm still handling dozens of requests for information weekly, all as a public service.
Pathology's modern founder, Rudolf Virchow M.D., left a legacy of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of all the make-believe and bunk that interfere with peoples' health, reasonable freedom, and happiness. I talk and write straight, and without apology.
Throughout these notes, I am speaking only for myself, and not for any employer, organization, or associate.
Special thanks to my friend and colleague, Charles Wheeler M.D., pathologist and former Kansas City mayor. Thanks also to the real Patch Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:

My home page
More of my notes
My medical students
Especially if you're looking for information on a disease with a name that you know, here are a couple of great places for you to go right now and use Medline, which will allow you to find every relevant current scientific publication. You owe it to yourself to learn to use this invaluable internet resource. Not only will you find some information immediately, but you'll have references to journal articles that you can obtain by interlibrary loan, plus the names of the world's foremost experts and their institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review. If you are interested in complementary medicine, then I would urge you to visit my new Alternative Medicine page. If you are looking for something on complementary medicine, please go first to the American Association of Naturopathic Physicians. And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I do not accept donations, though I appreciate those who have offered to help.
During the eighteen years my site has been online, it's proved to be one of the most popular of all internet sites for undergraduate physician and allied-health education. It is so well-known that I'm not worried about borrowers. I never refuse requests from colleagues for permission to adapt or duplicate it for their own courses... and many do. So, fellow-teachers, help yourselves. Don't sell it for a profit, don't use it for a bad purpose, and at some time in your course, mention me as author and William Carey as my institution. Drop me a note about your successes. And special thanks to everyone who's helped and encouraged me, and especially the people at William Carey for making it still possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!

![]()
![]()
|
|
|
|
|
|
|
|
|
|
|
|
|
![]() KCUMB Students
KCUMB Students
"Big Robbins" -- Endocrine
Lectures follow Textbook
QUIZBANK
Endocrine (It's impossible to separate pituitary, adrenal, thyroid, parathyroid, etc. Look at it all now.)
Mention the normal gross and microscopic anatomy of the adrenal glands, parathyroid glands, and thymus gland. Describe their origins within individuals, and their functions.
Define hypoadrenocorticism, mention the etiologies of the chronic and acute forms, and tell what each looks like clinically. Explain hyperpigmentation in some of these patients, and tell why they are at risk for sudden death.
Describe the etiologies of Cushing's syndrome, from the most to the least common. Tell what symptoms and signs should alert you, the physician, to the possibility of Cushingism. Explain Nelson's syndrome, and why it is becoming uncommon.
Define primary hyperaldosteronism and Conn's syndrome. Distinguish these from secondary hyperaldosteronism. Tell what symptoms and signs point to excess aldosterone, and explain the danger of treating these patients with "safe" diuretics.
Describe in detail the pathogenesis of congenital adrenal hyperplasia, and distinguish the most common salt-retaining and the most common salt-wasting form. Describe the forme fruste that we now believe is very common.
Describe the behavior of carcinomas of the adrenal cortex.
Discuss pheochromocytoma and neuroblastoma with respect to their names, locations, etiologies, catecholamine production, gross and microscopic appearances, clinical picture, and prognosis. Mention the "primitive neuroectodermal tumors" that look like neuroblastomas, and describe "spontaneous cures" of neuroblastoma. Provide an educated guess of how many of your classmates had a "neuroblastoma" at birth.
Describe in some depth the prevalence, etiologies, symptoms, signs and treatment of hyperparathyroidism. Explain how to tell parathyroid hyperplasia from parathyroid adenoma, and why anyone cares. Describe how and when hypoparathyroidism develops, why it is serious, and how to recognize it.
Describe how the size of the thymus gland changes with age. Define thymic hyperplasia and thymoma, tell what they look like, and mention the diseases with which they are associated.
List the components of the important anti-oncogene deletion syndromes MEN I, IIa, and IIb.
MAINTAIN A HIGH INDEX OF SUSPICION FOR ENDOCRINE DISEASE. This lecture ought to scare you.
|
|
|
One surprising fact about the adrenal gland is that, unlike many other organs, masses found here are seldom biopsied prior to excision. The radiology team will advise surgeons whether to remove particular masses. The one exception is biopsy to confirm metastatic disease in someone with a known cancer, usually in the lung (Arch. Surg. 144: 465, 2009).
THE ADRENAL CORTEX: "An organ essential to life." Pathology of the adrenal cortex: Arch. Path. Lab. Med. 132: 1263, 2008.
|
{11204} adrenal and its nerve, normal
{11207} adrenal and its nerve, normal {11210} adrenal and its nerve, histology, normal {15035} normal adrenal gland, showing zones (can you figure them out?) |
|
Unlike the pancreas, it isn't obvious where the "head", "body", and "tail" of an adrenal is located. The head is medial, the tail lateral.
The cortex, of course, has three "zones":
(1) ZONA GLOMERULOSA: mineralocorticoid production. Thin and patchy, small cells. (Most sources today say that ACTH does not affect the zona glomerulosa or mineralocorticoid production.)
(2) ZONA FASCICULATA: glucocorticoid production (now seems settled), resting cells (the reserve cells are at the ZG-ZR interface). Yellow.
(3) ZONA RETICULARIS: glucocorticoid production, androgen and estrogen production, grossly darker than outer layers. Brown.
(Mnemonic: Salt, sugar, and sex: the deeper you go, the sweeter it gets.)
Under the microscope, the borders are fairly easy to see.
In newborns, the future adult cortex is a thin layer under the capsule, and most of the gland is "fetal zone". This regresses in a few weeks and is usually gone altogether by the first birthday.
Future pathologists:
The normal adult weight of each adrenal gland is 4 gm.
If an adrenal gland weighs 6 gm or more (without a tumor), it is usually hyperplastic. The stress of the final illness increases the weight of the adrenals, which is why "normal autopsy weight" of an adrenal is sometimes given at 6-8 gm. Most violent suicides have adrenals weighing 9-11 gm each (Am. J. Psych. 144: 1214, 1987; confirmed AJFMP 19: 72, 1998.) The fetal adrenal, if enlarged, warns of impending premature labor.
* More for future pathologists: A bit of ectopic marrow, a few pigmented cells, or theca cells (especially in menopausal women) are normal.
* Ask a physiologist about the role of dehydroepiandrosterone in health; about 30% of a man's androgens are derived from this, and 90% of a post-menopausal woman's estrogens.
{49431} hyperplasia of adrenal cortex, etiology undisclosed
{09217} adrenal cortical hyperplasia, etiology unknown
* Around 2% of folks have adrenals with black nodules, usually without any evidence of dysfunction.
CONGENITAL ADRENAL HYPOPLASIA
Two types, both uncommon:
(1) Anencephalic type: thin cortex, no fetal zone; no ACTH during development
(2) Cytomegalic type: thin cortex composed entirely of large, bizarre cells (foci of such cells are common in normal newborns but regress).
It now turns out that a forme fruste allele at the DAX1 locus causes adrenal insufficiency in boys (no adults yet), i.e., this is a common, previously-unrecognized cause of adrenal insufficiency (J. Clin. Endo. Metab. 91: 3048, 2006).
Both present as hypoglycemic seizures in infants. Glucocorticoid replacement saves these children's lives.
ECTOPIC ADRENAL CORTICAL TISSUE (sometimes ectopic adrenal medulla too)
This is most common in the capsule ("capsular extensions") and at the origin of the celiac artery, but it can occur anywhere in the retroperitoneum, or under capsules of liver, kidney, ovary, or testis.
Ectopic adrenal caused problems when surgical adrenalectomy was very popular. ("A new adrenal gland grew back in a different place....")
| HYPOADRENOCORTICISM ("Addisonism", etc.): Insufficient glucocorticoid (and usually insufficient mineralocorticoid) production. Reviews Lancet 361: 1881, 2003; NEJM 360: 2328, 2009. |
|
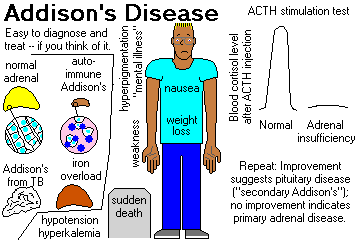
CHRONIC HYPOADRENOCORTICISM (ADDISON'S DISEASE, now regardless of etiology). Troubles start when 80% of the gland tissue is gone. Review Br. Med. J. 312: 1085, 1996.
You need to know the etiologies of chronic hypoadrenocorticism:
INFECTIONS
Most of Dr. Addison's patients had bovine TB![]() of the adrenals (by way of the lymphoid tissue of the
duodenum).
of the adrenals (by way of the lymphoid tissue of the
duodenum).
{09223} adrenal tuberculosis, gross
{25399} tuberculosis of adrenal, histology
{27257} tuberculosis of adrenal, histology
|
![]() TB of the adrenal
TB of the adrenal
WebPath Photo
Worldwide, fungal infections (remember histoplasmosis![]() ,
coccidioidomycosis
,
coccidioidomycosis![]() and South American blastomycosis)
and leprosy
and South American blastomycosis)
and leprosy![]() are important
causes, and now AIDS is too.
are important
causes, and now AIDS is too.
AUTOIMMUNE
The most prevalent non-iatrogenic cause of Addison's disease in the US today. The adrenal remnants are typically loaded with lymphocytes, etc., etc. Jack Kennedy suffered from this illness in his youth, and it was missed for several years (JAMA 201: 115, 1990). Even today, "delayed diagnosis of adrenal insufficiency is common": Am J. Med. Sci. 339: 525, 2010 ("twenty percent suffered for more than five years" -- especially when the principal complaint is "just not feeling well" / "low subjective health status").
![]() Jack Kennedy, second from left
Jack Kennedy, second from left
PT109 reunion, 1944
Notice weight and pigmentation
Long-mysterious, it's now clear that most of these patients have autoantibodies against 21-hydroxylase (Lancet 339: 1559, 1992). These are now generally called "adrenal cortex antibodies." Presumably this is antibody-dependent cell-mediated cytotoxicity, as in Hashimoto's disease.
And around 1.5% of people with type I diabetes make these antibodies, with progression to disease determined by genetic factors (J. Clin. Endo. Metab. 97: 1573, 2012).
Autoimmune adrenalitis often occurs jointly with Hashimoto's thyroiditis, type I diabetes mellitus, vitiligo ("autoimmune polyendocrine deficiency syndrome II", "polyglandular failure type II"; "Schmidt's syndrome", etc.)
Schmidt's is only the best-known of the autoimmune polyendocrinopathies in which there are various immune and non-immune disturbances tending to run together.
* There are probably other multi-antuoimmune diseases that are being worked out; consider endocrine disease when a patient with vitiligo or prenicious anemia seems unwell.
The more common Schmidt's (autoimmune polyglandular syndrome type II) is officially diagnosed when there is autoimmune addisonism plus either autoimmune thyroid disease or autoimmune diabetes (Am. Fam. Phys. 75: 667, 2007).
* Autoimmune polyglandular syndrome type III is diagnosed when there is autoimmune thyroid disease plus one or more other autoimmune diseases other than addisonism or hypoparathyroidism (Am. J. Med. Sci. 333: 178, 2007; J. Clin. Endo. Metab. 97: E1043, 2012). This is likely to change as these illnesses are further defined by molecular biology.
IATROGENIC
This results from too-rapid withdrawal of glucocorticoid medication, post-adrenalectomy for breast cancer or Cushingism, etc., ketoconazole or fluconazole antifungal drug therapy (Crit. Care Med. 29: 668, 2001), removal of a "non-functioning adenoma" (rare).
CORTICOSTEROID INSUFFICIENCY OF CRITICAL ILLNESS is a newly-characterized entity, seen especially in severe systemic infections, in which the body does not put out enough glucorticoid to handle the extra stress. The pathology's not worked out yet, but the impact on clinical practice in the ICU will be huge, since one must know whether to give supplementary cortisol. Consensus statements from the American College of Critical Care Medicine: Crit. Care Med. 36: 1937, 2009. See also Chest 135: 181, 2009.
OTHERS: Worth remembering are
{25394} adrenal cortical atrophy (key says "hypoplasia", I doubt this)
{24607} adrenal amyloidosis, gross
{15960} cytomegalic inclusion disease![]() , adrenal
, adrenal
{37216} adrenal leukodystrophy ("Lorenzo's oil") case, gross brain
{37218} adrenal leukodystrophy case, gross brain
{37221} adrenal leukodystrophy case, histology brain
{37224} adrenal leukodystrophy case, gross adrenal
{37225} adrenal leukodystrophy case, histology adrenal
* NOTE: Hollywood is Hollywood. The aftermath of "Lorenzo's oil" was disillusionment. It failed controlled studies miserably (NEJM 329: 745 & 801, 1993; NEJM 330: 1904, 1994; Ann. Neuro. 34: 121 & 169, 1993), and poisoned platelets (NEJM 328: 1126, 1993; Am. J. Hem. 44: 290, 1993; J. Inh. Metab. Dis. 17: 628, 1995) and (at least sometimes) natural-killer lymphocytes (J. Inh. Metab. Dis. 18: 101, 1995). We've now got two series of dead adrenoleukodystrophy patients who were treated in life with Lorenzo's oil. The scientific community pointed out that "Lorenzo's oil" doesn't even cross the blood-brain barrier, which is probably why it didn't work (Neuroch. Res. 19: 1073, 1995; Ann. Neuro. 36: 741, 1995). Yet another massive failure: J. Neurol. Neurosurg. Psych. 67: 290, 1999. However, interest continued. A study from Hopkins (Arch. Neuro. 62: 1073, 2005) that claimed success in preventing lesions in asymptomatic boys had only historical controls and also included other dietary alterations. Since the disease has a variable course in members of the same family, it's impossible to tell how much of the benefit was related to the treatment.
ACTH DEFICIENCY ("secondary hypoadrenocorticism")
These patients have almost always lost their adenohypophysis and have "panhypopituitarism". (Treat the whole person.... Caring for a little pituitary dwarf? Don't get focused on the height so that you forget the likely adrenal insufficiency.... J. Clin. End. Metab. 81: 1693, 1996). Less often, they have selective, presumably autoimmune, loss of the ACTH-producing cells: Arch. Int. Med. 152: 1705, 1992.
Clinical picture:
"Addisonian" patients show weakness, nausea, and weight loss, and are usually hypotensive (* 110/70 or less) and have other complaints. Like most endocrine patients, the problems are likely to appear "emotional".
In primary hypoadrenocorticism, the skin and buccal mucosa will usually be hyperpigmented, due to increased ACTH (MSH?) -- also look at freckles, nipples, palmar creases, old scars.
Lab studies typically show hyponatremia, hyperkalemia, metabolic acidosis, hypoglycemia, low serum cortisol, low urinary 17-OH-steroids, and (most important) failure to respond to various "stimulation tests" by increasing cortisol output.
It is common for these patients to die suddenly and unexpectedly before anyone thinks of adrenocortical insufficiency. This still happens (Br. Med. J. 312: 1085, 1996).
* Osteoporosis is severe in post-menopausal women with Addisonism, because of loss of adrenal androgens.
Replacement therapy is life-saving. (And get your patient a syringe of cortisol and an ID bracelet.)
{09371} Addison's disease; pigmentation and vitiligo (mother and daughter)
{09372} Addison's disease, face
{09373} Addison's disease, buccal pigmentation
{49438} Addison's disease, pigmentation
{49439} Addison's disease, pigmentation
{49440} Addison's disease, atrophy of the adrenal gland
SELECTIVE HYPOALDOSTERONISM is rarely due to primary disease of the adrenal cortex. (Clinicians talk about "hyporeninemic hypoaldosteronism".)
Much more often, the problem is really that the JGA is not producing renin (REE-nin, remember?). Usually the problem is diabetic arteriolar disease (no surprise); less often, it is one of the diseases of the renal tubules and/or interstitium.
* These patients have type IV renal tubular acidosis, exhibit normal response to ACTH stimulation testing, and need a prescription for oral 9α-fludrocortisone.
ACUTE HYPOADRENOCORTICISM ("adrenal apoplexy", "Addisonian crisis"): Sudden collapse, often fatal (the mechanisms are not fully understood, but it involves opening of the peripheral vasculature and shock with high cardiac output; consider giving any such patient glucocorticoid: Arch. Surg. 128: 673, 1993.) The same has been noted to keep blood pressure up in the brain-dead who are being maintained for organ removal (Anesthesiology 112: 1204, 2010). Update on how giving a little extra cortisol to all major trauma patients seems to help: JAMA 305: 1201, 2011.
PLEASE DO NOT MISS THIS ONE. It still gets missed: Lancet 385: 576, 2015.
It may result from undiagnosed adrenal insufficiency (iatrogenic, or patients stressed by infection, surgery, or treatment of concurrent myxedema; see for example J. Traum. 32: 94, 1992), or from known Addison's disease when extra glucocorticoids are not provided during stress.
WATERHOUSE-FRIDERICHSEN SYNDROME ("adrenal apoplexy") features hemorrhage, fibrin thrombi, and sometimes necrosis in the adrenals in a setting of sepsis. It's not clear whether death is due to adrenal shutdown, but it's not helping.
This occurs when there is overwhelming sepsis with hemorrhage into, and destruction of, the adrenals. Patients develop purpura, shock, and die in a few hours.
The etiologic agent is classically the meningococcus, though
staphylococci![]() (possible new
WF-producing strain NEJM 353: 1245, 2005), pneumococci, and H.
influenzae are other important causes (J. Clin. Path. 57: 208, 2004).
(possible new
WF-producing strain NEJM 353: 1245, 2005), pneumococci, and H.
influenzae are other important causes (J. Clin. Path. 57: 208, 2004).
W-F is not rare, and is often overlooked. One group suggests that if your patient in shock does NOT have elevated serum cortisol, he or she presumably has W-F. Draw blood, then give 200 or 300 mg of hydrocortisone (West. J. Med. 150: 582, 1989). Another protocol, for anybody who's septic: Am. J. Med. 98: 266, 1995.
We are now recognizing adrenal insufficiency in very-low-birth-weight preemies as a cause of circulatory collapse. Salivary cortisol screens for this and a bit of glucocorticoid supplementation helps a lot (J. Clin. Endo. Metab. 97: 890, 2012; Ped. Clni. N.A. 58: 1083, 2011).
{24606} Waterhouse-Friderichsen adrenal, gross
{09224} adrenal hemorrhage, consistent with Waterhouse Friderichsen
{07570} adrenal hemorrhage, gross, consistent with Waterhouse-Friderichsen syndrome
|
CUSHING'S SYNDROME: too much glucocorticoid. Review NEJM 332: 791, 1995.

1. IATROGENIC (the most common cause nowadays, preventable in part by giving "alternate-day" glucocorticoid therapy). Of course, the adrenals will be atrophic if glucocorticoids were administered, hyperplastic if ACTH was administered.
2. ACTH-PRODUCING PITUITARY LESION, usually a basophilic microadenoma ("Cushing's disease", "pituitary Cushingism")
The adrenals will usually be diffusely enlarged, but may be nodular, often with one or more large nodules (CT scanners take note -- a single "adenoma" does NOT necessarily rule out the need for pituitary surgery).
"Nelson's syndrome" -- rapid enlargement of the pituitary adenoma leading to hyperpigmentation, blindness and death -- followed adrenalectomy in many of these patients. (Why? It still happens -- sometimes the only way to relieve Cushingism is to remove the adrenals.)
3. ADRENAL CORTICAL ADENOMA OR CARCINOMA ("adrenal Cushingism"); the tumor may be primary, or an autonomous adrenal tumor may develop after years of "pituitary Cushingism")
4. ACTH- (OR CRH-) PRODUCING CANCERS OF OTHER ORGANS: oat-cell carcinoma (very well-known), bronchial and thymic carcinoids (rather common; Ann. Thor. Surg. 94: 1823, 2012; Mayo Clin. Proc. 69: 594, 1994; Ann. Thor. Surg. 95: 1797, 2013), medullary thyroid carcinoma, islet cell cancer; other APUDomas. Full-blown Cushingism is rare in oat cell patients, only because they don't live long enough.... Again, the hyperplasia is usually diffuse but may be nodular.
{49441} looks like an oat cell case; adrenal cortex is hyperplastic, and bears a metastasis
* Urocortin is a hormone widely distributed in the nervous system, with CRH-like activities; it is presently in search of a disease. Both CRF and urocortin are potent anorectic agents. The newly-discovered "urocortin 3" is also called "stresscopin". The truly hardcore can see J. Clin. Endo. Metab. 90: 4671, 2005.
5. Really "primary" adrenal hyperplasia (not due to excess ACTH):
* Genetic syndrome with too many cortisol receptors, low plasma cortisol: J. Clin. Endo. Metab. 85: 14, 2000.
* 6. Cushingism with a burst of cortisol after eating: inappropriate expression of GIP receptors on the adrenal cortex / adrenal adenoma (NEJM 327: 974, 1992; J. Clin. End. Metab. 81: 3168, 1996; J. Clin. Endo. Metab. 86: 583, 2001). There are other aberrant receptor problems as well: J. Clin. Endo. Metab. 88: 416, 2003.
* 7. Recurrent cushingism of pregnancy: Nobody understands it; the adrenal cortex must over-respond to some non-ACTH hormone made during gestation J. Clin. End. Metab. 81: 15, 1996; Clin. Endo. 54: 277, 2001.
Both Cushing's disease and glucocorticoid-secreting adenomas are most common in women ages 15 to 45, but can hit anybody, anytime. (* Cushingism in kids and teens: NEJM 331: 629, 1994).
Symptoms and signs that should alert you to possible Cushingism:
{09367} Cushingism, face
{09370} Cushingism, face
{16109} Cushing's syndrome
{16110} Cushing's syndrome
{16112} Cushing's syndrome "before"
{16111} Cushing's syndrome "after"
{49426} Cushingism, 40 y/o patient
{49427} Cushingism
{49428} Cushingism, hyperplastic adrenal cortex
* Future pathologists: Heavy negative feedback on pituitary basophilic ACTH-producing cells produces "Crooke's hyaline change".
PRIMARY HYPERALDOSTERONISM ("low-renin hyperaldosteronism"): too much mineralocorticoid (review: Postgrad. Med. 95(4): 199, March 1994; NEJM 339: 1820, 1999; Lancet 353: 1341, 1999; Surg. Clin. N.A. 84: 887, 2004; Lancet 371: 1921, 2008)
This results from "idiopathic" adrenal hyperplasia, or an adrenal adenoma.
This is important as a cause of surgically-correctable high blood pressure. Maybe 0.5% of hypertensives have primary hyperaldosteronism. The most recent work (Lancet 2008) uses the retrospectoscope to show that Conn's is probably not so common as has recently been claimed, but still an important concern.
Classically, patients exhibit hypokalemia, alkalosis, and low renin, and a failure of plasma aldosterone levels to increase significantly when the patient goes from supine to standing position.
Surprisingly, these patients do not have edema. (The effects of aldosterone in hanging onto body salt is overridden by atrial natriuretic peptide.)
Low potassium is likely to cause muscle weakness, and even paralysis.
Trap: These patients can die from hypokalemia if you give them thiazide diuretics to treat their high blood pressure.
Today, we screen by looking for the plasma aldosterone / plasma renin activity ratio. Some hypertensives have elevated levels, and many of these people will indeed have an aldosteronoma that can be removed (Am. J. Med. Sci. 324: 227, 2002); the rest will usually have hyperplastic cortices.
The most familiar cause is an "autonomous" adrenal cortical adenoma (CONN'S SYNDROME), often very small. It produces aldosterone (rare Conn-omas produce DOC instead). You'll clinch the diagnosis by sampling aldosterone levels in the adrenal veins (J. Clin. End. Metab. 86: 1066, 2001). Surgery is curative (Ann. Surg. 219: 347, 1994; Postgrad. Med. 95(4): 199, Mar. 1994); it is now routinely done via laparoscope (review J. Urol. 169: 32, 2003), and those that look harmless on scan may even be removed by partial adrenalectomy (J. Urol. 184: 28, 2010).
* We're just starting to sort out the somatic mutations that drive these tumors. Those with mutated CTNNB1 are super-powerful aldosterone producers (NEJM 373: 1429, 2015.)
For "bilateral adrenal hyperplasia", medical treatment is the norm, or if sampling the adrenal veins reveals one gland to be producing much more aldosterone than the other, removing that gland usually works (J. Am. Coll. Surg. 213: 106, 2011).
![]() Adrenal cortical adenoma
Adrenal cortical adenoma
Produced Conn's
Wikimedia Commons
The rest of the patients have "idiopathic hyperaldosteronism", with normal or hyperplastic adrenals. These patients get spironolactone. Not surprisingly, the borderland between these and the adenomas is blurry (Surgery 106: 1161, 1990); probably it's best to operate only if the hypertension is unsuppressible medically like a Conn-oma.
A few patients have glucocorticoid-correctable hyperaldosteronism and hypertension. This is transmitted autosomal-dominant. It is now clear that the problem is a chimeric beta-hydroxylase/aldosterone synthase gene (Nature 355: 262, 1992; Lancet 339: 1024, 1992; screening kids Arch. Dis. Child. 71: 40, 1994; J. Urol. 154: 510, 1995). Update J. Clin. Endo. Metab. 87: 3187, 2002. When the cell is told by ACTH to make cortisol, it pumps out huge amounts of aldosterone, too. (Thinkers: Giving a tiny amount of exogenous glucocorticoid solves the problem. How? If you can't answer this, go back and review your endocrine physiology.)
* Hypertension from a mutated aldosterone receptor stuck in the "on" position: Science 289: 119, 2000.
* Another cause is "apparent mineralocorticoid excess syndrome", a lack of 11-β-hydroxysteroid dehydrogenase type 2, which turns cortisol to cortisone in the renal tubules; cortisol ends up overstimulating the mineralocorticoid receptors. The forme fruste may be a common contributor to "idiopathic" low-renin hypertension even with normal potassium. See J. Clin. Endo. Metab. 86: 1247, 2001; Lancet 353: 1341, 1999. Yet another is a 21-deoxyaldosteronoma (J. Clin. End. Metab. 80: 737, 1995). Rarely an ovarian cancer produces aldosterone (series Arch. Int. Med. 156: 1190, 1996).
SECONDARY ALDOSTERONISM is much more common. It is part of the picture in CHF, cirrhosis, nephrotic syndrome, Goldblatt hypertension, and other common problems.
Don't forget Bartter's hypokalemia (vessels are insensitive to angiotensin and/or the sodium pump in the ascending loop of Henle doesn't work -- Hosp. Pract. 29(5): 103, 1994.)
Don't confuse this with salt-retaining congenital adrenal hyperplasia (see below).
CONGENITAL ADRENAL HYPERPLASIA: autosomal-recessive virilization syndromes that, in their most severe forms, affect young children.
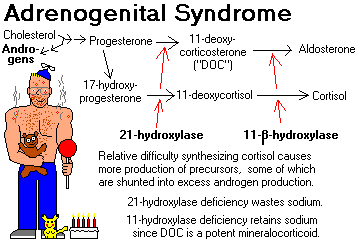
Deficiencies (mild or severe) of the various enzymes required to synthesize cortisol result in decreased production of cortisol and other hormones.
This results in increased ACTH, with resulting adrenal cortical hyperplasia.
Steroid precursors are shunted into the production of abnormally large amounts of the androgen androstenedione (ambiguous genitalia in girls, "infant Hercules" and Leydig cell nodules in boys, etc.)
Remember these two types (there are at least six others):
21-HYDROXYLASE DEFICIENCY (most common): no cortisol, aldosterone, or DOC, hence salt wasting. This gene is inside the HLA locus. Review J. Clin. Endo. Metab. 88: 2624, 2003.
11-BETAHYDROXYLASE DEFICIENCY: huge amounts of DOC, causing salt retention and high blood pressure (molecular biology of the gene Proc. Nat. Acad. Sci. 90: 4552, 1993).
Full-blown congenital adrenal hyperplasia is a devastating illness, especially for women. Mild variants of these syndromes (i.e., relatively ineffective enzymes -- especially 21-hydroxylase deficiency) are probably widespread -- causing, for example, amenorrhea in girls or hirsutism in older women.
It's important to find these people because a little dexamethasone given daily will greatly improve the internal milieu.
To test your female patient with amenorrhea or hirsutism, administer ACTH and measure plasma 17-hydroxyprogesterone one hour later. It will be elevated if your patient has even mild 21-hydroxylase deficiency.
{49437} adrenogenital syndrome 2 year old girl
{24450} adrenogenital syndrome, virilized baby girl
{49432} 11-hydroxylase deficiency, 11 month old boy
ADRENAL CORTICAL ADENOMAS
These are round, yellow (like the adrenal cortex) nodules. (* Purists call them "nodular hyperplasia" if the surrounding cortex is at all lumpy-bumpy).
Adrenal cortical adenomas are surprises at around 2% of autopsies and abdominal CT scans. They can cause Cushing's syndrome, Conn's syndrome, or virilization -- but the vast majority seem to produce nothing.
They are commonly discovered on CT scans too, and clinicians are learning to ignore small adrenal masses ("incidentalomas", so long as there is no evidence of steroid or catecholamine over-production).
Consider screening folks with incidentalomas for Cushingism. Maybe 20% of these really are active, and contribute to hypertension, diabetes, obesity, and osteoorosis, even if the patient is not floridly Cushingoid. Common sense triumphant. See Surg. Clin. N.A. 84: 875, 2004. "Occult/subclinical Cushingism due to incidentaloma" is now a recognized entity: J. Clin. Endo. Metab. 88: 5808, 2003. NIH Consensus Conference: Ann. Int. Med. 138: 424, 2003.
A question that is NOT settled and is interesting is whether removing a little adrenal cortical incidentaloma will relieve "features of mild hypercortisolisim" (i.e., when the dexamethasone suppression test / late-night salivary cortisol levels / 24 hour urinary cortisol don't show real Cushingism -- but your patient is still fat / glum / hypertensive / glucose intolerant). See J. Clin. Endo. Metab. 95: 4106, 2010.
As above, adrenal masses are generally not biopsied prior to excision. The case against fine-needling incidentalomas, made by surgeons: Surgery 142: 497, 2007.
* Leave discussions of the arcana of these common lesions to us. This includes "spironolactone bodies" (pink scroll-like things, also found in the ZG of folks taking spironolactone), "black adenomas", and much more.
"Adrenal cysts" seen on scans have unpredictable pathology: Cancer 101: 1537, 2004.
{09220} adrenal cortical adenoma, gross
{20312} adrenal cortical adenoma, gross
{49436} adrenal cortical adenoma, gross; this one produced Conn's syndrome
{10298} adrenal cortical adenoma
{20315} adrenal cortical adenoma, histology
{09221} adrenal cortical adenoma, histology
{09222} adrenal cortical adenoma, histology
{08964} adrenal cortical adenoma, histology (hard to tell from normal cortex)
{09052} adrenal cortical adenoma, electron micrograph; note tubular cristae in mitochondria
(spaghetti instead of lasagna)
{09375} effect of masculinizing adrenal cortical adenoma, "before"
{09374} effect of masculinizing adrenal cortical adenoma, "after"
{49434} gynecomastia in five-year old boy, feminizing adrenal cortical adenoma
|
|
ADRENAL MYELOLIPOMA: a metaplasia-choristoma made of bone marrow. They can be big but are generally harmless. You can see bone marrow in adrenal cortical hyperplasia too.
{25412} adrenal myelolipoma, gross
{49443} adrenal myelolipoma, gross
{25413} adrenal myelolipoma, histology
![]() Adrenal myelolipoma
Adrenal myelolipoma
Pittsburgh Pathology Cases
ADRENAL CORTICAL CARCINOMA (Am. J. Clin. Path. 105: 76, 1996; J. Urol. 169: 5, 2003; J. Clin. Endo. Metab. 95: 4812, 2010)
These are rare cancers that are often lethal. Many are are hormonally active and produce Cushing's, Conn's, and/or virilization.
Future pathologists: These are usually obviously malignant, grossly and microscopically, with ten or more mitotic figures per 10 high power fields.
The most reliable way of spotting malignancy is still the mitotic figure count; other systems have not proved very reliable (Am. J.Clin. Path. 127: 398, 2007).
* The molecular biology is just starting to be understood. About 90% of these tumors have overexpressed IGF-II.
* Mixed endocrine syndromes usually mean cancer. Adrenal tumors that feminize, or that produce androgens without glucocorticoids, are most often malignant.
Today's surgeons are operating these cancers in the hope of cures, and there are enough cases now with good results that it's possible to demonstrate the value of taking lymph nodes (Ann. Surg. 255: 363, 2012) and of removing single lung metastases (Ann. Thorac. Surg. 92: 1965, 2011).
Mitotane, an analogue of the old-fashioned insecticide DDT, is the classic mainstay of therapy. (It will also destroy and scar any remaining normal adrenal gland.)
* Future pathologists: We used to use the electron microscope to find tubulovesicular mitochondria, which are characteristic of adrenal cortex to confirm that something was really an adrenocortical carcinoma. SF1 stain ("steroidogenic factor 1") is sensitive and specific (J. Clin. Endo. Metab. 95: E161, 2010) for adrenal cortical carcinoma (really, for origin in a steroid-producing cell) if you are in doubt. Criteria for malignancy have been developed; since this cancer is not particularly treatable, their usefulness is limited.
{24087} adrenal cortical carcinoma, gross
{40196} adrenal cortical carcinoma
{24090} adrenal cortical carcinoma, histology
NOTE: Cancer in the adrenals is usually metastatic carcinoma. Half of all lung cancers eventually metastasize to the adrenals. Adrenal insufficiency sometimes results when replacement is massive, but is usually missed clinically.
![]() Metastatic cancer in the adrenals
Metastatic cancer in the adrenals
WebPath Photo
![]() Cancer metastatic to the adrenal
Cancer metastatic to the adrenal
AFIP
Wikimedia Commons
THE ADRENAL MEDULLA: "An organ not essential to life".
Around 10% of the normal adrenal by weight. The source of "adrenalin" (epinephrine, also norepinephrine). At autopsy it is gray, unless it has autolyzed. (The old name "adrenal capsules", from the pre-refrigeration era, reflects the fact that the medulla had usually liquified by the time the anatomists and pathologists got to it.)
"Adrenal medullary hyperplasia" (i.e., the medulla is too big; one way to tell is that it extends into the tail, where it's not supposed to go) is a marker for MEN II and a few rarities. It can be diffuse or nodular, and it may be best to call any nodule bigger than 1 cm a pheochromocytoma.
The only bona-fide diseases are two tumors that may arise here or at the other chromaffin tissue masses -- pheochromocytoma (well-differentiated, adults) and neuroblastoma (poorly-differentiated, children).
* The story of the old adrenal-to-brain transplant for parkinsonism: Mayo Clin. Proc. 65: 305, 1990.
PHEOCHROMOCYTOMA ("paraganglioma", "pheo", formerly "ten percent tumor"; big NIH consensus review Ann. Int. Med. 134: 315, 2001; big review for pathologists Arch. Path. Lab. Med. 132: 1272, 2008).
This tumor is named for its colorful reaction in fixatives containing chromic acid salts.
Pheochromocytomas secrete norepinephrine (most common) and/or epinephrine (usually less, * and often other things, including dopamine, serotonin, ACTH, somatostatin, neuropeptide Y, and/or VIP; Cancer Res. 49: 7010, 1990).
Pathologists confirm that a likely-looking tumor is a pheochromocytoma by staining it up for chromogranin and/or synaptophysin.
The infamous paroxysms of extreme hypertension, accompanied by sweating, headache, and other autonomic disturbances, probably result from physical compression and/or ischemia of the "pheo".
Even a tiny (1 gm) benign pheochromocytoma can make a person very sick and will eventually cause death.
Today, "pheochromocytoma" is defined to arise in the adrenal medulla. Similar tumors (less common) arising elsewhere are called "extra-adrenal paragangliomas." These sites include the organs of Zuckerkandl ("para-aortic bodies", i.e., the little nubbins of chromaffin tissue at the origin of the inferior mesenteric artery and/or aortic bifurcation -- prominent and surely important in the unborn child, but regressed by birth), paravertebral sympathetic chain, urinary bladder (patients get terrible headaches whenever they urinate), or "paraganglia" such as the carotid body.
The old business about "ten percent of pheo cases involve both adrenals, 10% are familial, and 10% metastasize" is history. Today, everything's about the new genetics.
* "Composite pheo" contains some neuroblastoma, ganglioneuroblastoma, or nerve-sheath tumor; think of MEN II or neurofiromatosis. If there's no neroblastoma, the presence of other elements is probably not a bad prognostic indicator. Update Am. J. Clin. Path. 132: 69, 2009.
You will often be reminded of the MEA ("MEN", "multiple endocrine adenoma/neoplasia syndromes") -- common autosomal dominant conditions that predispose patients to certain endocrine tumors. Pre-natal diagnosis is available for these tumor-gene syndromes. Learn them now:
MEN I: PPP (Wermer's syndrome), gene MENI, protein menin * on 11q13; * less often the gene CDKN1B
Parathyroid adenoma(s) (1 or, often, more glands) / "chief cell hyperplasia" (i.e., all four glands): NEJM 321: 213, 1989).
Pituitary adenoma (anterior)
Pancreatic islet cell adenoma (gastrinoma)
* The pituitary-and-parathyroid-only variant usually is from a different locus that remains to be discovered: J. Clin. Endo. Metab. 92: 1948, 2007.
MEN IIa: PAC (Sipple's syndrome); the RET gene (Nature 367: 315, 375, 377 & 378, 1994; NEJM 335: 943, 1996; J. Clin. End. Metab. 81: 2711, 1996; screening for the gene J. Clin. Endo. Met. 78: 1261, 1994 and Mayo Clin. Proc. 72: 430, 1997; new alleles keep appearing J. Clin. Endo. Metab. 89: 4142, 2004; surveillance J. Clin. End. Metab. 82: 897, 1997).
Parathyroid adenoma(s) (1 or, often, more glands) / "chief cell hyperplasia"
Adrenal medullary tumor (pheochromocytoma) or hyperplasia
Calcitonin-producing hyperplasia-carcinoma of thyroid
MEN IIb (MEN III; * eponyms like Gorlin-Vicker haven't caught on):
Similar to MEN IIa; the patients have Marfanoid body habitus and mucosal (ganglio)neuromas (bumps on the edges of their tongues and elsewhere), and are less likely to have parathyroid problems. Same locus, different allele (Nature 1994, see above.)
Grossly, pheos are very bloody (because they are very vascular), and often show fibrosis, calcification, cystic change, or even * fatty change (?!)
Microscopically, pheos resemble adrenal medulla.
Special stains are available that show norepinephrine and/or epinephrine in granules (* future pathologists: aqueous fixation washes them out.)
* Nowadays, a tumor that stains for phenylethanolamine-N-methyltransferase (PNMT) has the "adrenergic phenotype", one that doesn't has the "noradrenergic phenotype".
Of course, that's history now. Pheos light up with chromogranin A and tyrosine hydroxylase. Extra-adrenal paragangliomas light up with chromogranin A but often not with tyrosine hydroxylase.
For pheos, the traditional teaching has long been that there are no histologic criteria for malignancy, not even vascular invasion. The honest pathologist cannot predict the tumor's behavior.
To prove malignancy you must find pheo in a location where it could not have arisen. Five-year survival rate with malignant pheo is around 50%.
* "Pheo balls" are hyaline spheres that can be very big. You can see them in most normal medullas if you look hard enough. They must be thanatosomes. Fun to know: They are acid-fast and autofluorescent!
* A pheo or neuroblastoma, as a frozen section, exposed to formalin, fluoresces yellow-green from the catecholamines getting altered. This is a helpful study that pathologists can perform immediately on retrieval of the tumor.
Regardless of location and appearance, the patients will report anxiety, headache, palpitations, "panic attacks", sweating, dizziness, etc. (Again, you may suspect the basic problem is emotional. "Pheo is a great imitator.")
"Textbook" pheochromocytoma patients have paroxysms of severe hypertension. Actually, the majority show sustained high blood pressure.
Pheochromocytoma is present in fewer than 1% of people with high blood pressure, but it's a diagnosis you don't want to miss.
Pheos are still often missed clinically (Am. J. Surg. 179: 212, 2000) and are all-too-familiar surprises at autopsy (Lancet 335: 1189, 1990). The patients typically had been told they had "benign essential hypertension" and "emotional problems".
In addition to causing bad high blood pressure and all that goes with it, high levels of circulating catecholamines can directly (and likely permanently, as with cocaine) damage the myocardium can cause coronary spasm, and can play havoc with smooth muscle (renal arteries, bowel, brain, etc. -- angiographers may report "vasculitis".)
Screening tests for pheos detect increased amounts of catecholamines or their metabolites in blood or urine.
The classic screen for pheos (24 hour urinary vanillylmandelic acid -- VMA) is being superseded by more sensitive and specific tests.
Today's "most sensitive screen" is the plasma free metanephrine assay (Ann. Int. Med. 134: 315, 2001; Arch. Int. Med. 160: 2521, 2000; JAMA 287: 1427, 2002).
There's a radioisotope scanner/treatment for pheo and neuroblastoma -- I-131 labeled metaiodobenzylguanidine (MIBG); it's not very sensitive for diagnosis (J. Clin. Endo. Metab. 86: 685, 2001). Today, the 6-[18F]fluorodopamine PET scan seems to be preferred for spotting metastatic pheochromocytoma.
* Finding the hidden pheo using novel techniques, including 6-[18F]-fluorodopamine: J. Clin. Endo. Metab. 86: 3641, 2001.
* The Mayo crew examines how much it would cost to screen every hypertensive patient for pheo using each of three methods, and simply states that spending $50,000 or $100,000 per patient found isn't worth it (J. Clin. Endo. Metab. 89: 2859, 2004); I am not sure I agree, and certainly you need to work up the young ones, the ones with headaches, and the ones with "nerves".
Treatment is surgical, with very careful management of fluid status and blood pressure before and after surgery (J. Urol. 161: 764, 1999). The anesthesiologist, of course, has an extra challenge (Anesth. Analg. 91: 1118, 2000). And surgeons must be careful manipulating the tumor! Adrenal sparing surgery, i.e., let's leave a bit of cortex behind: Br. J. Surg. 86: 94, 1999. Today, it's likely that the tumor will be removed successfully via the laparoscope (Urol. Clin. N.A. 28: 97, 2001; update Arch. Surg. 145: 893, 2010).
* Injecting epinephrine to fake a pheo: JAMA 266: 1553, 1991 (weird!).
{20316} pheochromocytoma in adrenal, gross
{25417} pheochromocytoma, gross
{49444} pheochromocytoma, gross
{09226} pheochromocytoma, gross
{09227} pheochromocytoma, showing positive brown staining with chromic acid ("chromaffin")
{08874} pheochromocytoma, histology
{08873} pheochromocytoma, histology
{25418} pheochromocytoma, histology
{09228} pheochromocytoma, histology
{09229} pheochromocytoma, histology, positive chromaffin reaction
{09080} pheochromocytoma, electron micrograph showing granules
{09081} pheochromocytoma, electron micrograph showing granules
{08056} pheochromocytoma cardiotoxicity
{08053} pheochromocytoma cardiotoxicity; note contraction bands
NEUROBLASTOMA (Ped. Clin. N.A) 55: 97, 2008; genes JAMA 307: 1062, 2012)
Either Wilms tumor or neuroblastoma is the most comon extracranial solid cancer of children. It is derived from primitive nerve elements (* and the cells will always grow neurites, at least in tissue culture). Discovered by Virchow.
A majority of neuroblastomas arise in or near the adrenals.
They seem to strike at random. * Three risk loci, all at 6p22, have been spotted (NEJM 358: 2585, 2008); an autosomal dominant allele of ALK produces neuroblastoma when it amplifies itself (Nature 455: 930, 2008).
Grossly, neuroblastomas are soft, white tumors.
Portions often undergo dystrophic calcification (which helps the radiologist make the diagnosis.)
The tumor eventually metastasizes widely. "Blueberry muffin baby" is a repulsive, classic description for a neuroblastoma patient with multiple skin metastases.
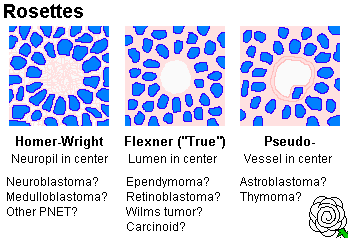
Histologically, neuroblastoma is a tumor of "small blue cells" (i.e., cancers with small, fairly-uniform cells with scanty cytoplasm). Often (but not always) the cells are arranged in "rosettes" (recalling neural tubes) around a tangle of neurites (* "Homer Wright rosettes", pretty-much diagnostic). EM shows neurosecretory granules and often neurites too. (* True "Flexner" rosettes surround a hole. Think of ependymomas or retinoblastomas.)
* Future pathologists: Immunohistochemistry helps differentiate this from other "tumors of small blue cells" (the LEMON family), and will also help you recognize their nondescript cells in bone marrow. The better-differentiated neuroblastomas are likely to stain positive for neuron specific enolase, S-100, and/or synaptophysin, but the more primitive ones will be negative for everything. In tissue culture, neuroblastoma cells sprout neurites almost at once.
Grading of the tumor is based on mitotic figure counts, with karyorrhexis being taken into account nowadays also: Cancer 77: 1582, 1996 updates the older Shimada system which was based on how much stroma was present and whether the cells were maturing (i.e., differentiating as neurons with more cytoplasm.) The update is the "International Neuroblastoma Pathology Committee" system (Cancer 86: 364, 1999; Cancer 92: 2451 & 2699, 2002; Cancer 94: 1574, 2002). "MKI" is the "mitosis-karyorrhexis index", i.e., what percent of cells exhibit these; more than 4% is bad, less than 2% is good. The systems for distinguishing "favorable" and "unfavorable" histopathology now incorporate stroma, pattern, and maturation as well as the child's age but MKI is still central (J. Clin. Onc. 27: 289, 2009).
Most neuroblastomas produce catecholamines, resulting in elevated urinary metabolites. (* They also produce certain characteristic protein markers, etc., etc.)
The classic test involves checking urine for homovanillic acid (HVA) and vanillylmandelic acid (VMA). Japan screens all babies at six months of age (Lancet 2: 152, 1988); skeptical Brits suggest that they find only regressing tumors (Lancet 337: 344, 1991). The Germans have screened their babies at one year and find it lot of cases, but I couldn't tell whether they're saving lives (J. Clin. Onc. 17: 1200, 1999 -- anybody want to do a lab presentation on this?). Now the Japanese are doubting whether they've saved any lives, either (Canc. Caus. Contr. 9: 631, 1998; "we haven't" J. Ped. Surg. 37: 949, 2002). Neuroblastoma screening has not caught on in the US in the "managed care era"; both big studies indicated it's a bad idea NEJM 346: 1041 & 1047, 2002.
There are also a variety of curious (probably autoimmune) paraneoplastic syndromes that result from neuroblastomas, including neurodegenerative disorders similar to those in oat-cell carcinoma.
Prognosis in neuroblastoma:
And of course, it's good if the cells are differentiating...
In a baby, the tumor is likely to regress/differentiate/mature to a "differentiating neuroblastoma" (more than 5% of the cells look like big ganglion cells, and/or some schwann-cell-like stroma), a stroma-rich GANGLIONEUROBLASTOMA (a neuroblastoma, probably with lots of big ganglion cells, with 50% or more being a stroma that looks like schwann cells) or a thoroughly benign GANGLIONEUROMA; Br. J. Surg. 83: 263, 1996). Ganglioneuromas are made of ganglion-like cells in a fibrous "schwannian" stroma; it may occasionally appear in adults (J. Clin. Endo. Metab. 95: 3118, 2010; Surgery 147: 854, 2010; Surgery 149: 99, 2011). In maybe 2% of autopsies on infants dying of unrelated causes, there is a neuroblastoma-like "incidentaloma". Obviously most of these cure themselves. We wish we knew exactly why/how this happens.
* The process begins with the appearance of S100-positive Schwann cells, which are from outside the tumor. If the tumor is to self-cure, it must be near-triploid and have intact chromosome 1 (NEJM 334: 1505 & 1537, 1996).
{24716} neuroblastoma, histology, good rosettes
{25420} neuroblastoma, gross
{25422} neuroblastoma, histology
{39049} neuroblastoma, gross; probably an incidental finding in a newborn
{09009} neuroblastoma, histology
{09232} neuroblastoma, histology
{20046} neuroblastoma, histology
{20047} neuroblastoma, histology
{09011} neuroblastoma, histology, good rosettes
{08963} neuroblastoma histology (sorry, no good rosettes)
{25424} ganglioneuroblastoma, histology
{25426} ganglioneuroblastoma, histology
{24608} ganglioneuroma, gross
![]() Neuroblastoma patient & family
Neuroblastoma patient & family
Cindie and 10 y/o Derek Madsen
Pulitzer-Prize photoessay
In toddlers, spontaneous remission is less likely, but even metastatic disease is often cured by chemotherapy.
In older kids and adults, neuroblastomas grow slower but seldom self-cure or respond well to therapy (Cancer 79: 2028, 1997). We are getting good results even in the more difficult cases nowadays thanks to I131-131-MIBG (Cancer 117: 4286, 2011).
Retinoblastomas (cones of the eye), medulloblastomas (cerebellum), pinealoblastomas (pineal), and adult neuroblastomas (lots of places -- many of these are probably Ewing variants) are related pediatric tumors that look like neuroblastomas microscopically.
* The tendency today is to call these "primitive neuroectodermal tumors", despite obvious differences in their basic biology.
* Neuroblastoma of the olfactory epithelium is a special entity. Don't worry about it.
INTRODUCTION TO ADRENAL TESTING
The aphorism -- "A physician is only as good as his/her index of suspicion" -- is especially applicable to endocrine disease. As an alert clinician, you will often suspect adrenal gland problems and will want to order sensitive tests.
Some classic cases:
If the morning serum cortisol is >13 mcg/dL, you are probably not dealing with addisonism. If the morning serum cortisol is <=13 mcg/dL, order a rapid ACTH ("cosyntropin", "synacthen") stimulation test.
After 30-60 minutes, the serum cortisol should spike to at least 500 nanomoles/Liter = 18 micrograms/dL.
If it doesn't, you have adrenal insufficiency, either primary (the glands are diseased) or secondary (the glands atrophied from lack of ACTH stimulation).
In the past, we used to give an ACTH stimulation test each day for a week; patients with secondary adrenal insufficiency would grow their adrenals back from the test (and probably want to come back for more ACTH!)
Nowadays, ACTH plasma assays are good enough to make the distinction easier. Injected ACTH clears from the blood in a few minutes. Nowadays, use the 30-minute blood sample to assay not just cortisol, but also serum ACTH (it'll probably still be high in primary adrenal disease, low-ish in pituitary insufficiency). Also check serum aldosterone in the same sample; the ability of the adrenals to produce aldostone isn't lost as much as the ability to produce cortisol when the problem is in the pituitary gland. A lack of a normal spike after ACTH administration confirms primary adrenal insufficiency.
A "spot serum cortisol" is worthless (the stress of venipuncture can cause a false negative.) A "24 hour urine cortisol" is worthless (it can be zero in health.) A "spot serum ACTH" is worthless. It's secreted in pulses.
Order serum cortisol determinations at 8 AM and 8 PM (circadian rhythm is always lost in Cushingism), plus a low-dose dexamethasone suppression test, OR just order one (or maybe two or three) 24 hr urinary free cortisol assays. The most convenient screen now being promoted is a late-night salivary cortisol (J. Clin. Endo. Metab. 94: 456, 2008).
Screen with a serum aldosterone/renin ratio. If high, measure urine aldosterone on a high-salt diet AND/OR see whether you can suppress the aldosterone using fludrocortisone AND/OR check to see if plasma aldosterone fails to increase on standing up AND/OR consider performing a saline infusion aldosterone-suppression test AND/OR consider a CT scan. Nobody really knows what's best. Current thinking: J. Clin. Endo. Metab. 91: 2618, 2006.
Administer ACTH and measure blood 17-OH-progesterone.
Check serum/urine catecholamines and/or metabolites, ask your lab.
Of course, only some of these patients are endocrine cases. As a rule, meaningful hormone assays are performed under conditions of attempted stimulation (if you suspect deficiency) or attempted suppression (if you suspect over-production). If your screening tests are positive, or if you have any doubts, get consultation.
If you diagnose endocrine disease that is not present, the patient gets lifelong medication, unnecessary surgery, or unnecessary radiation. If you fail to diagnose a disease that is present, the patient is likely to die of a disease that might have had an excellent prognosis. (Suicide is common among patients with untreated Cushing's syndrome.) If you make the correct diagnosis, the treatment of endocrine disease is very satisfying to physician and patient alike -- because it works.
|
|
|
![]() Parathyroids
Parathyroids
"Pathology Outlines"
Nat Pernick MD
The parathyroids arise from the third and fourth branchial clefts. They are each 3-4 mm across and weigh maybe 35 mg each (there is no consensus about "ideal total weight"). A parathyroid gland 6 mm or more across is too big.
* They were first discovered by Richard Owen in 1850 in a rhinoceros in the London zoo. Virchow supposedly noticed in humans; they were named by a Swedish medical student.
Future surgeons: The key to telling a parathyroid gland / tumor at surgery is that (unlike fat, thyroid nubbins, or lymph nodes), a droplet of blood will ooze up when the gland is pricked by your scalpel blade. Why?
Textbooks show four. Most people have 3 or 4 parathyroids, less often 5 or even 6.
Unusual locations (especially for the lower pair) are common and explainable embryologically.
"Ectopic" parathyroid glands may be found in the carotid sheaths, behind the esophagus, in the anterior mediastinum, in the pericardial sack, thyroid gland, etc. The new sestamibi scan has made finding these much easier.
Cysts are uncommon but do occur (J. Otol. 18: 311, 1990).
Cell types in the normal glands:
Chief cells: typical hormone-secreting endocrine cells; they are easily stained for PTH
Oxyphils ("oncocytes"): large, pink-staining cells that appear after puberty and occur in clusters. (By EM, these are seen to be packed with mitochondria, like Hürthle cells. They do not contain secretory granules, and the mitochondria are probably not metabolically active. Oxyphils are more numerous in older people.)
"Water-clear cells" ("wasserhelle" cells): seen in some parathyroids. (They are full of glycogen, and their functional status is uncertain.)
Fat cells: especially after puberty. "Fatty ingrowth", if you like. Except in the rare "lipoadenoma", all tumors and hyperplasia of the parathyroid have diminished fat.
Parathyroid hormone is the major regulator of calcium homeostasis in humans.
Its production-secretion is regulated by serum calcium levels.
The N-terminal assay measures the active hormone, though the form measured by the C-terminal portion stays around longer. (Pitfall: the C-terminal portion is filtered through the kidneys, and is increased when the kidneys are underfunctioning even if parathyroid function is normal.)
Historically, I've suggested measuring both. With high PTH, high calcium, and low phosphorus, you have your diagnosis. Today's i-PTH (intact PTH) might be more expensive but is available stat for use during surgery. Other labs aren't so useful. (Did you know that urinary cyclic AMP is a good estimate of parathyroid hormone levels?)
Effects:
Kidney: Promotes conversion of 25-OH-D3 to the very active 1,25-OH-D3 that increases calcium absorption from the gut.
Promotes resorption of calcium from the glomerular filtrate, and promotes loss of phosphate.
This effect is mediated by activation of adenylate cyclase. Serum parathyroid levels are accurately reflected by measuring urinary c-AMP concentrations.
Bone: Promotes resorption of calcium by osteoclasts (via activation of adenylate cyclase.)
Increased levels of parathyroid hormone causes proliferation of osteoclasts. (Finding of an osteoclast in a section of non-pagetic, non-injured bone from an adult usually means hyperparathyroidism.)
Gut: Promotes calcium absorption (indirect effect, via vitamin D activation).
HYPERPARATHYROIDISM ("stone and bone disease"; review Mayo Clin. Proc. 77: 87, 2002; pathologists see Arch. Path. Lab. Med. 134: 1639, 2010)
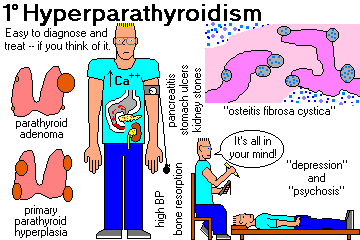
PRIMARY HYPERPARATHYROIDISM: Due to disease of the parathyroid glands. Pathologists see Arch. Path. Lab. Med. 132: 1251, 2008; NEJM 365: 2389, 2011).
80-85%... parathyroid "adenoma" ("single gland disease")
10-15%... parathyroid "hyperplasia" ("multiple gland disease", usually all four glands)
1-3%... parathyroid carcinoma (I think this traditional number is high)
<1%... iatrogenic seeding causing lots of little glands: Hum. Path. 21: 234, 1990, (* by my teacher, Dr. David Roxe); Surgery 116: 111, 1994.
Some people may also use this term to include hypercalcemia caused by production of parathyroid-hormone-like activity (PTH-rP) by cancers, notably squamous lung cancer), or very rarely real PTH. Today the term "pseudohyperparathyroidism" is preferred.
Primary hyperthyroidism is a common clinical problem. About 1 person in 1000 will need parathyroid surgery during his or her lifetime. When serum calcium was added to the chemical profiler in 1974, a huge number of patients were found and much good was done by removing their tumors.
Calcium rises because of enhanced GI absorption and renal reabsorption. There is also a tiny contribution from increased osteoclastic activity (Am. J. Med. Sci. 320: 334, 2000).
Symptoms and signs:
Elevated serum calcium on routine screening (probably the commonest presentation today; nowadays if the albumin-adjusted calcium is even a little bit high and the serum parathyroid hormone isn't low, you've got the diagnosis made)
Mental changes (depression, psychosis; Am. J. Med. 101: 111, 1996)
Kidney stones (the commonest presentation in the past)
Nephrocalcinosis (metastatic calcification in the tubular basement membranes with eventual damage to the tubules)
Bone changes (first osteomalacia, then widespread involvement of the skeleton by increased osteoclastic activity especially in the centers of trabeculae; finally cystic lesions variously known as "osteitis fibrosa cystica", "von Recklinghausen's disease of bone", or "brown tumors." All this heals after the hyperparathyroidism is fixed.)
Most extreme: Calciphylaxis (metastatic calcification of the skin, with horrible ulcers and breakdown): South. Med. J. 102: 318, 2009.
|
|
Just not feeling right. Hypercalcemia makes you fatigued before anything else. People who eventually turn out to have primary hyperparathyroidism are much more likely to have been missing a lot of work "for being sick" for several years prior to diagnosis (BMJ 317: 848, 1998.) After surgery, "asymptomatic" people with parathyroid adenomas feel a lot better (Surgery 128: 1013, 2000).
Resorption of the tufts of the phalanges on x-ray
* "Band keratopathy" of Bowman's membrane in the cornea
* Loss of lamina dura on dental x-rays
Gastric ulcers (5%; hypercalcemia from any cause enhances gastrin secretion)
Hypertension (50%, cured by parathyroid surgery only if the kidney is undamaged)
Pancreatitis (occasionally)
Pseudogout (occasionally)
* NMJ problems (denervation-atrophy picture on muscle biopsy)
* Skin ulcers (following metastatic calcification of vessels: Arch. Path. Lab. Med. 114: 482, 1990).
Labs:
high serum calcium
low serum phosphate
high 24-hour urine calcium excretion (i.e., you absorb a lot more through your gut)
high urinary cAMP (old-fashioned test)
high serum parathyroid hormone (PTH) for your screening
PARATHYROID ADENOMA
The commonest cause of primary hyperparathyroidism.
* In the mid-1980's, there was a fad notion that these all represented "nodular hyperplasia". Newer genetic studies have shown these tumors truly are monoclonal. Well usually. And often the hyperplasias are clonal. It's blurry.
These tumors are most common in older women, but may occur in anyone. They average around a gram but are sometimes bigger.
The basic biology very often involves translocations that deregulate the oncogene PRAD-1 (bcl-1, cyclin D1, a cell-cycle regulator; Nature 350: 512, 1991, much more since).
* When I first begin preparing these notes in the 1980's, I mentioned that I'd seen several at autopsy that were probably non-functional. In the scanning era, surgeons are now making the same discovery (Surgery 138: 1111, 2005).
The surgeon finds three normal glands and an adenoma that is easily removed (10% are in an "ectopic location.)
In the late 1990's, parathyroid surgery was revolutionized by the introduction of the Tc99 sestamibi scan to locate the lesion(s) preoperatively, and intraoperative radioguidance (Arch. Surg. 135: 481 & 550 & 844 & 1461, 2000; Ann. Surg. 23: 31 & 331 732, 2000; J. Am. Coll. Surg. 190: 540, 2000; Surg. Clin. N.A. 80: 1399, 2000; Surgery 129: 720, 2001; Arch. Surg. 137: 659, 2002).
* Imprint cytology of the parathyroid and other neck structures at surgery: Arch. Path. Lab. Med. 127: 64, 2003; Am. J. Clin. Path. 118: 895, 2002.
The adenoma often has a rim of compressed normal gland at the edge. Most adenomas contain few if any adipocytes, and most of the cells do not have intracytoplasmic fat.
An adenoma can be composed of any of the three kinds cells (chiefs, oxyphils, waterclears).
As you would expect, parathyroid adenomas (even in people with no family history) often lack 11q13 (the MEN-I locus).
* Every once in a while, there is so much fat in a parathyroid adenoma that it looks like a lipoma, i.e., the infamous "lipoadenoma".
* Only the very largest parathyroid adenomas (more than 3.5 grams) are likely to result in hypoparathyroidism after removal (the other glands were suppressed and are atrophic): Surgery 154: 718, 2013.
{10827} parathyroid adenoma, histology
Nowadays, some folks talk about "just following" people with a parathyroid adenoma who doesn't have symptoms (Am. J. Med. 124: 911, 2011). I would demand surgery. If the serum parathormone level is more than three times the upper limit of normal, or there is a palpable neck mass, it's likely to be cancer and I don't think anyone would question the need to operate (Am. J. Surg. 202: 590, 2011).
PARATHYROID CARCINOMA (Cancer 100: 900, 2004; Surgery 142: 936, 2007; J. Clin. Endo. Metab. 96: 3678, 2011)
A rare cause of primary hyperparathyroidism.
By definition, this cancer arises in a parathyroid gland and produces parathyroid hormone. It tends to be slow-growing, and recurs in about half of people after it is resected.
A familial syndrome of parathyroid adenomas and carcinomas plus jaw tumors involves mutant HRPT2 (parafibromin: NEJM 349: 1722, 2003); this gene is not mutated in parathyroid adenomas, either familial or sporadic (J. Clin. Endo. Metab. 90: 5015, 2005).
* Loss of Rb, reportd in the 1990's, was a disappointment; loss of heterozygosity for this and other anti-oncogenes is noted in some tumors showing bizarre mitoses, invasion, or metastases (Surgery 144: 949, 2008); we await studies with adenomas as controls.
* Today, the "other" marker stain that indicates malignancy in a parathyroid tumor is PGP9.5 (J. Clin. Endo. Metab. 94: 434, 2009).
These cancers are somewhat aggressive. About a third are cured with simple excision, another third recur and require re-operation for cure, and only a third eventually metastasize and ultimately cause death, usually from refractory hypercalcemia. Sestamibi scanning has greatly improved the management of these tumors (Clin. Nuc. Med. 32: 358, 2007).
* Please leave the distinction among "parathyroid carcinoma", "atypical parathyroid adenoma", and "parathyromatosis" to us pathologists. Thanks. Cancer 110: 255, 2007.
PARATHYROID HYPERPLASIA
The second most important cause of primary hyperparathyroidism. All four glands are big, for no obvious reason. Even though this is "a different disease from adenomas", the masses are often clonal, and (in the case of chief-cell hyperplasia) the same genes put you at risk.
This may occur in anyone, but is suspicious for one of the multiple endocrine neoplasia (multiple endocrine adenomatosis) syndromes. See below.
Hyperplastic glands usually lack the usual fat cells.
CHIEF CELL HYPERPLASIA is the common kind. It raises the possibility of MEN I or MEN II. Older techniques to tentatively distinguish a hyperplastic gland from an adenoma on morphology have fallen into disuse.
WATER-CLEAR HYPERPLASIA is a different disease with the same symptoms. All four glands are quite big, probably because there is much non-functioning cytoplasm in the clear cells. This time, there is no intracellular lipid but plenty of glycogen.
{27260} parathyroid hyperplasia (arrow sign helps)
{09271} primary parathyroid hyperplasia, histology
|
|
|
In parathyroid hyperplasia, the bulk of the parathyroid tissue must be removed, leaving a small amount behind. (Sometimes a small amount of parathyroid tissue gets transplanted to the forearm, for future whittling.)
NOTE: The honest pathologist CANNOT distinguish a hyperplastic gland from an adenoma! The surgeon MUST send samples of two glands. (Why? Confirmed yet again.... Surgery 142: 930, 2007) The pathologist will report "hypercellular parathyroid tissue". We can tell the second (normal) gland easily using a touch-prep cytology.
SECONDARY HYPERPARATHYROIDISM: Parathyroid hyperplasia due to hypocalcemia (or hyperphosphatemia, or hPTH resistance, or vitamin D deficiency, or vitamin D resistance) from some other cause, usually renal failure (less often malabsorption or malnutrition).
In people who are vitamin D deficient, the extra parathyroid hormone keeps calcium levels normal (J. Clin. Endo. Metab. 85: 4125, 2000; Am. J. Med. Sci. 319: 380, 2000; remember there's plenty of this in the US); serum parathormone levels are part of the screen especially by epidemiologists.
Less common causes are intestinal malabsorption, calcitonin-producing tumors (i.e., medullary carcinoma of the thyroid) and rickets (low serum phosphate, in contrast to renal failure, in which serum phosphate levels are high.)
Serum calcium is low-normal.
Bone disease (as in primary hyperparathyroidism, but now called "renal osteo-dystrophy") is a big problem. Some patients may need partial parathyroidectomy to control it.
Yesteday's patient with secondary hyperparathyroidism of renal origin was managed with calcitriol (Am. J. Med. Sci. 320: 100 & 107, 2000). Cinacalcet, a medication that activates the calcium sensors on the parathyroids, is the mainstay of today's treatment and the regression of the previously-enlarged parathyroids is well-documented, though for some patients it does not work and there is a characteristic lesion (J. Clin. Path. 64: 756, 2011).
TERTIARY HYPERPARATHYROIDISM: Hypercalcemia develops in a setting of secondary hyperparathyroidism.
One or more glands has "become autonomous" in the setting of secondary hyperparathyroidism / parathyroid gland hyperplasia, and overproduces parathyroid hormone. Probably this means it has lost the MEN-I anti-oncogene on chromosome 11 (J. Clin. End. Met. 76: 139, 1993). Usually we see this in patients with kidney failure, and more often than not it will regress after kidney transplantation -- though sometimes removal of an overactive gland is required (Am. J. Med. Sci. 339: 420, 2010). Or it may appear after secondary hyperparathyroidism is corrected by the transplant (Am. J. Clin. Path. 135: 100, 2011).
* NOTE: Genetic typing of parathyroid masses has helped us recognize that the above scheme, while a bit simplistic, is fundamentally accurate. If one gland is involved, it's an adenoma and will show one of two different genetic profiles. If two, three, or four glands are involved, it's "multiple gland parathyroid neoplasia", with different genetic signatures for each. See Am. J. Path. 165: 565, 2004.
OTHER HYPERPARATHYROID SYNDROMES:
In the 1970's, many of these folks were operated in search of parathyroid disease... sorry!
The mutation is in CaSR, the calcium sensing receptor (Clin. End. 50: 537, 1999; Am. J. Hum. Genet. 64: 189, 1999), which tells the nucleus what the plasma calcium level is. Parathyroid hormone is overproduced as a result. The alleles and how they produce disease are well-studied (J. Clin. Endo. Metab. 95: E245, 2010); having the gene checked is now a part of the workup of any less-than-obvious case of hypercalcemia (J. Clin. Endo. Metab. 95: 1819, 2010). More alleles J. Clin. Endo. Metab. 98: E1692, 2013.
* Two doses gives neonatal severe primary hypercalcemia requiring total parathyroidectomy.
* Other mutations give a familial hypocalcemia (J. Clin. Endocrin. Metab. 84: 363, 1999), i.e., in these "activating" mutations, the receptor is stuck in the "on"-position. An autoantibody that causes the same problem: NEJM 351: 362, 2004. Mutations of the G-protein subunit 11 (G11) produce a familial hypocalciuric hypercalcemia (loss of function) or an autosomal dominant hypocalcemia (gain of function; NEJM 368: 2476, 2013).
Order a calcium-creatinine clearance ratio, which will be very low (less than .01) in these patients.
JANSEN'S METAPHYSEAL CHONDRODYSPLASIA, an autosomal dominant syndrome caused by an overactive PTH1R parathyroid hormone receptor. Hypercalcemia and short-limbed dwarfism. Worked out NEJM 335: 708, 1996.
* Inability to break down activated vitamin D: mutated CYP24A1 (NEJM 36: 401, 2011.)
HYPERCALCEMIA: Differential diagnosis for beginners. Review Postgrad. Med. 115: 69, 2004.
HYPOPARATHYROIDISM
The most common cause is iatrogenic (following thyroid surgery). This is a complex subject and still fairly common (Br. J. Surg. 97: 1687, 2010).
* A high-tech device that picks up the near-infrared signal from the calcium sensor in the parathyroid glands, so thyroid surgeons can know where they are and leave them behind (!! Surgery 154: 1371, 2013).
Next is autoimmunity ("polyglandular failure type I", the syndrome with addisonism, ectodermal dysplasia, and mucosal
candida![]() ? --
J. Clin. Endo. Metab. 88: 4602, 2003).
In both familial and sporadic cases, the autoantigen is often CaSR (J. Clin. Inv. 97:
910, 1996; updates
J. Clin. Endo. Metab. 89: 4484, 2004; J. Clin. Endo. Metab. 94: 4655, 2009;
J. Clin. Endo. Metab. 98: 3884, 2013).
You also remember DiGeorge's syndrome.
? --
J. Clin. Endo. Metab. 88: 4602, 2003).
In both familial and sporadic cases, the autoantigen is often CaSR (J. Clin. Inv. 97:
910, 1996; updates
J. Clin. Endo. Metab. 89: 4484, 2004; J. Clin. Endo. Metab. 94: 4655, 2009;
J. Clin. Endo. Metab. 98: 3884, 2013).
You also remember DiGeorge's syndrome.
Kearns-Sayre mitochondrial myopathy / mitochondriopathy (J. Clin. Endo. Metab. 83: 125, 1998) for some reason produces hypocalcemia.
Wilson's, Riedel's struma; X-linked agenesis syndrome (J. Clin. Inv. 115: 2822, 2005); various genetic syndromes (for example J. Clin. Endo. Metab. 91: 4587, 2007).
Symptoms and signs of hypocalcemia begin with mental changes, circumoral paraesthesia, Chvostek's sign, Trousseau's sign, and progress to carpopedal spasm, convulsions, tetany. Check for cataracts, too; nobody knows why they tend to occur when ionized calcium is low.
The diagnosis is confirmed by finding low serum calcium and high serum phosphate.
* Treatment includes vitamin D and calcium gluconate cookies (lifelong, usually manageable) Injectable parathormone is now available (J. Clin. Endo. Metab. 88: 4214, 2003) and for refractory cases can be infused continuously by pump (J. Clin. Endo. Metab. 96: 3308, 2011; J. Clin. Endo. Metab. 97: 391, 2012).
By the way... there's plenty of vitamin D deficiency (low calcium, low phosphate) thanks to lack of sunlight (chosen lifestyles, veiling of women) and/or malnutrition (fad diets, poverty, malabsorption, stupidity) and/or vitamin D resistance (genetic syndromes). Update on hypocalcemia: BMJ 336: 1298, 2008.
PSEUDOHYPOPARATHYROIDISM
Some curious disorders. In each, there is an inability to carry the parathyroid hormone receptor signal to the cell's machinery. (Proc. Nat. Acad. Sci. 95: 10038 & 11798 & 15475, 1998; Am. J. Med. Genet. 77: 261, 1998; J. Clin. End. Metab. 81: 1660, 1996).
As you'd expect, serum calcium runs low, phosphate runs high, and (except in type II) urinary cAMP fails to rise in response to parathyroid hormone administration.
In Pseudohypoparathyroidism TYPE IA, there is a mutation of a portion of the GNAS1 gene that codes for Gsα. There is a striking imprinting effect (Am. J. Hum. Genet. 68: 1283, 2001.)
Hence, if you inherited a bad allele from your mother, you get resistance to parathyroid hormone (marked), plus some resistance to other endocrine hormones. Plus, you get the bony deformities ("Albright's hereditary osteodystrophy"), with short stature, short fingers, and a round face.
If you inherited a bad allele from your father, you get ONLY the bony deformities ("pseudopseudohypoparathyroidism"). Update on all of this stuff: J. Clin. Endo. Metab. 95: 651 & 765, 2010.
* Certain bad alleles inherited from Dad produce the thankfully-rare progressive osseous heteroplasia, in which skin and muscle transform into bone (NEJM 346: 99 & 128, 2002 -- now distinguished from true "myositis ossificans"). Alleles and molecular biology: J. Clin. Endo. Metab. 95: 3028, 2010.
In Pseudohypoparathyroidism TYPE IB, the bones are formed normally. The kidney is resistant to the effects of parathyroid hormone and there is milder resistance to TSH as in type Ia. The mutated gene (* STX16 / syntaxin 16) is adjacent to the GNAS1 locus; it's recently been identified as a portion of the complex that's responsible for its being imprinted (J. Clin. Inv. 1255: 112, 2003). It can only be expressed if it is inherited from Mom.
* In Pseudohypoparathyroidism TYPE IC, the alpha subunit is normal but there is the osteodystrophy. In the 1990's I predicted it was caused by a different allele at the GNAS1 locus and this has now been demonstrated (J. Clin. Endo. Metab. 87: 189, 2002; J. Ped. Endo. 19-S2: 635, 2006).
In Pseudohypoparathyroidism TYPE II, there is resistance to the effects of parathyroid hormone in bone and kidney, and some bone deformities. However, the urinary cAMP response to PTH challenge is normal, and The GNAS1 locus is not involved (i.e., there's a problem with the signal distal to cAMP). The genetics remains obscure.
* If you lack both copies of the real parathyroid hormone receptor, you die as a baby of a severe dyschondroplasia. Carriers are asymptomatic.
![]() Child with pseudo-pseudo-hypoparathyroidism
Child with pseudo-pseudo-hypoparathyroidism
Courtesy of Mary Fay MD
THYMUS
|
|
Originates from the third and (* ?) fourth pharyngeal pouches (* hence, the most common location for an ectopic or supernumerary parathyroid gland -- Am. J. Surg. 203: 292, 2012.)
Large in neonates, it starts to involute after puberty, and is usually just a mass of fat in older people.
The cortex (many lymphocytes, almost all T-cells) and medulla (few lymphocytes, mostly T-cells, some B-cells) have as their basic structural unit an unusual stellate epithelial cell.
* A few of these cells differentiate as keratinizing squamous pearls, or Hassall's corpuscles.
You are already familiar with the severe underdevelopment of the gland in such illnesses as ataxia-telangiectasia, DiGeorge / Nezelof, and some (not all) forms of severe combined immunodeficiency. Usually these people lack good corticomedullary differentiation and Hassall's corpuscles, as well as lymphocytes.
Thymic cysts are fairly common (3rd branchial pouch), but don't assume all cysts are benign -- for some reason, cysts often form near a thymus-involving cancer (Hodgkin's, germinoma).
* Ask a pathologist about the "myoid cells" in the medulla. They contain actin, myosin, and myoglobin. Maybe this has something to do with the whole myasthenia gravis connection.
{14760} normal kid's thymus; a=cortex, b=medulla, c=vessel
{13958} Hassall's corpuscles stained for keratin (this appears to be normal thyroid)
* Stress lesions in the thymus are of interest to pathologists, and help us tell how long somebody was seriously sick. Before 12 hours, there are no changes. At 12-24 hours, you start seeing macrophages eating lymphocytes. At 24-48 hours, you start seeing a starry sky. Over 48 hours, the corticomedullary junction becomes blurry as the gland atrophies. After 72 hours, the gland is atrophic and there will be no further changes.
FOLLICULAR HYPERPLASIA is said to present when there are large germinal follicles in the organ.
Most folks do have a few tiny germinal follicles in the thymus gland. I'd ask you to reserve the term THYMIC FOLLICULAR HYPERPLASIA for situations in which the gland itself is oversized. This is common in folks with various autoimmune diseases (especially lupus and addisonism), and in early HIV infection.
Patients with myasthenia gravis, an autoimmune diseases caused by anti-NMJ antibodies, also show thymic hyperplasia (unless it is removed or destroyed by a thymoma), and thymectomy is the best treatment for myasthenia gravis (probably even ifit is not hyperplastic: Ann. Thoracic Surg. 91: 212, 2011).
* The less-common form of myasthenia gravis, caused by antibodies against muscle-specific kinase, is not helped by thymectomy (Brain 126: 2304, 2003).
* True hyperplasia of the thymus is simply a gland that's oversized for the person's age. For some reason that no one understands, this is fairly common in adults who have been cured of a malignancy by chemotherapy.
THYMOMA is a histologically-benign tumor of the epithelial cells of the thymus gland. There are often lymphocytes mixed in, but these are non-neoplastic.
The gross appearance (i.e., whether or not the tumor invades the mediastinal structures) is most important for prognosis.
The World Health Organization (1999) classification of thymomas is now standard, and predicts outcome quite well (Ann. Thor. Surg. 77: 183, 2004); one group thinks it's too complicated and a simplified system would work just as well in answering the real question (i.e., who should get chemo / radiation? Cancer 112: 2780, 2008).
Not classified yet... Fibrosing variant: Am. J. Clin. Path. 121: 867, 2004.
A's can expect cures, AB's are usually cured (Ann. Thorac. Surg. 77: 1183, 2004). The rest are more dangerous, but the stage of the tumor at presentation is more important, and pathologists cannot distinguish the entities in type B very well: Chest 127: 755, 2005. And just to make the whole thing less helpful, more than half of thymomas have a mix of at least two different histologic types, and stage is much more important than grade (Am. J. Clin. Path. 137: 444, 2012).
{13952} thymoma, gross
{25653} thymoma, gross
{13955} thymic tumor, possibly Hodgkin's
{49097} malignant thymoma, gross
Among thymoma patients...
Myasthenia gravis occurs in around 50% (* and around 30% of myasthenia gravis patients have a thymoma; thymomas express acetyl choline receptor epitope on their cells Lancet 339: 707, 1992; update on these thymomas Ann. Thor. Surg. 79: 219, 2005).
Acquired "pure red cell aplasia" (stop putting out red cells, presumably autoimmune) occurs in around 20%
Hypogammaglobulinemia occurs in a few percent and is called Good's syndrome (rare, but in the "diff" of nearly everything)
Other thymus tumors:
LYMPHOMAS (T-cell, of course), CARCINOIDS / NEUROENDCRINE TUMORS(aggressive, and likely to produce ACTH or other troublesome hormones: Am. J. Clin. Path. 114: 200, 2000; Ann. Thor. Surg. 74: 133, 2003; the histology does not predict behavior Chest 124: 141, 2003; spotting them on needle aspiration Arch. Path. Lab. Med. 130: 1612, 2006; update Ann. Thor. Surg. 94: 241, 2012), * THYMIC CARCINOMAS (i.e., obviously malignant on histopathology: Cancer 67: 1025, 1991), and * germinomas (seminomas -- check for positive staining for placental alkaline phosphatase -- , less often embryonal-cell carcinomas or choriocarcinomas) arise in the thymus.
* Thymic TERATOMAS are relatively common.
* CASTLE -- "carcinoma showing thymus-like elements" arises in the thyroid but looks and stains like thymoma (Am. J. Surg. Path. 30: 994, 2006).
* There's a popular "five T's" mnemonic for anterior mediastinal masses:
* Patients with an enlarged thymus found on routine imaging can probably be followed without biopsy if they have no symptoms. Biopsy is probably indicated if there are symptoms, since the chances of finding a malignant lymphoma are fairly good (J. Thor. Card. Surg. 140: 977, 2010).
![]() Carcinoid
Carcinoid
Mediastinum
Pittsburgh Pathology Cases
* Brian Piccolo, of "Brian's Song" fame, had a thymic embryonal-cell type carcinoma, as did football player Dan Turk.
* "Status thymicolymphaticus" was an imaginary disease, dreamed up to explain SIDS and infanticide, for which several million newborns received radiation therapy. They are now at much increased risk for papillary carcinoma of the thyroid. (There's a lesson here -- self-deception is certain in the absence of proper controls.)
PINEAL ("the third eye"): Tumors of the pineal are troublesome because of their location. In children, pineal tumors are likely to produce sexual precocity.
PINEALOMA, except for its location, looks exactly like a seminoma or dysgerminoma.
Other germ-cell tumors occur in the pineal also, so you could see a pineal teratoma, a pineal choriocarcinoma, or any other. (I've seen all three.)
PINE(AL)OBLASTOMA (see above) resemble neuroblastomas and medulloblastomas.
PINEOCYTOMAS are made up of cells like those in adult pineal. Both are aggressive cancers seen most often in children.
GLIOMAS, etc., can occur in the pineal. Pineal tumors: Cancer 72: 870, 1993. Cysts: Neurology 41: 1034, 1991 (fooled me once).
{03998} normal pineal gland, anatomy
{02815} normal pineal gland, gross
{01223} normal pineal gland
{01239} normal pineal gland histology, with brain sand
{05219} pineal cyst, gross
{01711} pineal germinoma, gross
* THE MELATONIN BUSINESS: Older review Am. Fam. Phys. 57(8): 1783, 1998.
Since the stuff is dirt-cheap to make, occurs naturally, and isn't patentable, it's now widely available over the counter.
If you believed everything you heard about melatonin, you'd be reading uncritically. What is clear is that its effects on insomnia and jetlag have impressed people far more than most "supplement" stuff.
A meta-analysis found melatonin basically useless overall in sleep disorders, even jet lag and shiftwork disorder (for which it was once famous: BMJ 332: 385, 2006). Since the pineal often calcifies in older folks, and perhaps fails as a result, look forward to melatonin as a sleep aid especially in geriatrics. The most recent study showing an effect of the stuff showed it worked best on patients who don't secrete normal amount of melatonin metabolite, and that these were mostly older folks (Am. J. Med. 116: 91, 2004).
There are some hopeful results in irritable bowel syndrome (Gut 54: 1353, 2005) and a non-yet-reproduced report of improvement in tardive dyskinesia (Arch. Gen. Psych. 58: 1049, 2001). An optimistic study in children (Clin. Ped. 42: 51, 2003) lacked controls. For old folks with "cognitive decline, mood, behavioral and sleep disturbances", melatonin seems to help as long as it's also given with bright light (JAMA 299: 2642, 2008; I couldn't help but wonder if the old folks weren't simply being given more attention, hence the benefit.) For major depression: Lancet 378: 621, 2011.
In the meantime, the stuff seems extremely safe, and I wouldn't fault you for wanting to experiment, assuming you give proper informed consent.
A man said to the universe:
"Sir, I exist!"
"However," replied the universe,
"That fact has not created in me
A sense of obligation."
-- Stephen Crane
BIBLIOGRAPHY / FURTHER READING
I urge anyone interested in learning more about endocrine pathology to consult these standard textbooks.
In my notes, the most helpful current journal references are embedded in the text. Students using these during lecture strongly prefer this. And because the site is constantly being updated, numbered endnotes would be unmanageable. What's available online, and for whom, is always changing. Most public libraries will be happy to help you get an article that you need. Good luck on your own searches, and again, if there is any way in which I can help you, please contact me at scalpel_blade@yahoo.com. No texting or chat messages, please. Ordinary e-mails are welcome. Health and friendship!


| New visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if people like me who call ourselves Christians didn't try to act as good as other good people ." Prayer Request

| If you have a Second Life account, please visit my teammates and me at the Medical Examiner's office. |
Teaching Pathology
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |
 |
Click here to
see the author prove you can have fun skydiving without being world-class. Click here to see the author's friend, Dr. Ken Savage, do it right. |


 Endocrine
Endocrine

 Paragangliomas
Paragangliomas